采用SLAF-BSA技术分析玉米尾孢菌灰斑病抗性位点
2021-01-08张小飞
李 菁,张小飞
(1.西安文理学院 生物与环境工程学院,陕西 西安 710065;2.西安市农业技术推广中心,陕西 西安 710061)
玉米(ZeamaysL.)是粮食、饲料和工业原料兼用型作物,在农业生产和经济发展中占有重要地位。由于栽培品种遗传基础狭窄,加上多年的连作、气候变化等多重影响,每年由于病害造成的产量损失达总产的10%以上。玉米灰斑病是由尾孢菌(Cercospora)侵染引起的一种世界性玉米病害,1924年在美国伊利诺伊州首次被发现[1]。我国在辽宁、吉林、黑龙江等北方春玉米区、黄淮海夏玉米区普遍发生,在云南、四川、湖北等地发生呈现快速上升趋势,已经成为我国玉米生产上重要叶部病害,对玉米生产造成了很大威胁。玉蜀黍尾孢菌(Cercosporazeae-maydis)和玉米尾孢菌(C.zeina)为引起玉米灰斑病的2种主要病原。在辽宁、吉林、黑龙江、河北发生的灰斑病病原为玉蜀黍尾孢菌,在贵州、四川、陕西、河南发生的灰斑病病原为玉米尾孢菌[2]。由于玉米尾孢菌灰斑病是新发生病害,对抗病遗传的了解尚未深入,致使玉米尾孢菌灰斑病抗病基因定位进展较缓。对灰斑病的前期研究多集中在由玉蜀黍尾孢菌侵染的灰斑病,而对由玉米尾孢菌侵染的灰斑病抗性基因定位缺乏深入研究。
目前,基因定位主要是基于遗传图谱定位与集群分离分析法(Bulked segregant analysis, BSA)[3]。传统遗传图谱定位周期长、密度低、成本高;而BSA分析是将分离群体中表现出极端性状的个体混合起来构建2个混池,快速定位与目的基因紧密连锁分子标记的分析方法。SLAF-seq(Specific locus amplified fragments sequencing)又称为特异性位点扩增片段测序技术,是大规模基因分型的一项非常有效的方法,该技术具有诸多优点,包括标记开发成本低、效率高和大群体的高容纳力等,已广泛运用于功能基因定位、遗传图谱构建和遗传多态性研究[4-6]。
本研究选用玉米尾孢菌灰斑病抗病自交系R225与感病自交系掖478作为亲本,通过杂交、自交等方法构建F2分离群体共345个单株,在2013-2014年连续2 a采用田间自然诱发在云南德宏、湖北恩施进行抗灰斑病表型鉴定,利用集群分离分析法(BSA)对F2遗传分离群体(2个亲本和30株抗病材料+30株感病材料的极端性状混池)进行玉米尾孢菌灰斑病抗性关联分析,为今后开展玉米尾孢菌灰斑病抗性QTL的精细定位和分子标记辅助育种提供了材料基础。
1 材料和方法
1.1 材料及构建方法
根据多年多点的田间自然诱发辅以人工接种鉴定结果并参考农艺性状表现,选取玉米自交系R225和掖478为供试材料。R225是四川省农科院植保所玉米研究室连续多年筛选的高抗灰斑病的优良自交系,因绝大部分表现高抗或抗病的自交系属于热带或亚热带种质背景,而R225是为数不多的温带种质中的高抗自交系,表现高抗,高配合力,对发掘温带材料抗性基因具有重要意义。掖478为高配合力的感病自交系,是我国玉米育种研究工作中普遍采用的优势自交系,对玉米性状改良具有很高的育种应用价值。2011年冬季在海南三亚以R225为父本,掖478为母本进行杂交,获得F1(经抗性鉴定,植株均表现抗病),2012年夏季将F1植株套袋自交产生F2群体共345个单株,将亲本、F2分离群体于2012年冬播种于海南试验基地,并套袋自交产生F2:3家系种子。抗性鉴定于2013,2014年连续2 a在云南德宏、湖北恩施田间自然诱发表型鉴定,在玉米进入乳熟期开始调查。调查时目测每份鉴定材料群体的发病状况,记载病情级别。
1.2 叶片DNA提取、样品处理
当玉米生长至5-6叶期时,每株取少量新鲜叶片,采用CTAB法提取F2单株的基因组DNA,用NanoDrop 2000超微量分光光度计测定A260及A280,计算样品浓度,并以1%的琼脂糖凝胶进行电泳检测。经纯度和浓度检验合格后,选取30株抗病单株DNA样品进行等量混合,组成F2抗池(R混池),将30株感病单株DNA样品进行等量混合,组成F2感病池(S混池),将亲本与抗、感病池DNA用于后续SLAF-seq测序。
1.3 酶切方案的设计及SLAF标签分析
将玉米基因组作为参考基因组进行酶切位点预测并制定后续酶切方案,经检测提取的基因组DNA浓度及纯度合格后,对各基因组DNA样品进行酶切。将所得酶切片段(即SLAF标签)按照顺序分别进行3′端加A尾处理、Dual-index4测序接头连接处理、PCR扩增、产物纯化、样品混合、电泳选取目的片段进行切胶,待构建的SLAF-seq文库质量检测合格后,应用Illumina HiSeqTM 2500测序仪进行高通量测序。为评价酶切试验的有效性,同时选择水稻(Oryzasativa)作为对照样品(Control)进行同步测序,测序工作由北京百迈客生物技术有限公司完成。
1.4 信息分析流程
数据分析步骤参照王伟等[7]方法,利用Dual-index对所获得的原始测序数据进行识别,得到各个样品的读数(reads)。测序读数经过滤接头后对测序质量和数据量进行进一步的考察和评估。通过对照试验的数据来进行RsaⅠ+HaeⅢ酶切效率的评估,据此来判断试验过程是否符合准确性和有效性的标准。通过比对读数(reads)与参考基因组,在亲本和混池中开发SLAF标签,在亲本中搜寻具有多态性的SLAF标签和位于reads覆盖区域中的SNP位点。进一步将所得SNP位点进行关联分析,得到与抗性性状紧密相关的位点,并根据关联阈值确定候选区域,然后进一步对候选区域内的基因在GO、SwissProt、NR、COG及KEGG 5个数据库中进行功能注释。
2 结果与分析
2.1 酶切与建库
使用玉米基因组为参考来预测酶切位点,根据酶切原则选择了RsaⅠ、HaeⅢ 2种限制性内切酶。酶切片段在414~444 bp为SLAF标签。对SLAF标签在每条染色体上的数目进行统计,由表1可见,Chr1标签数最多为23 398个,Chr10标签数最少为11 678,与染色体长度正相关,染色体越长,标签数越多。10个染色体共计162 429个SLAF标签,可见SLAF标签在基因组各染色体上基本呈现均匀分布(图1)。说明酶切方案可行。

表1 各染色体中的SLAF标签数量

图1 SLAF标签在参考基因组上的分布
2.2 测序数据统计和检验
通过去除接头100 bp×2读取长度作为后续使用的数据进行评价和检验,以保证数据质量。对各样品的测序reads数量、Q30(即测序质量值≥30)和GC含量,共获得42.05 M reads数据,测序平均Q30为83.77%,平均GC含量为47.07%(表2)。水稻Control测序获得0.55 M reads的数据量。用SOAP软件将对照的测序reads与参考基因组进行比对,双端比对效率在80.90%,比对效率正常。
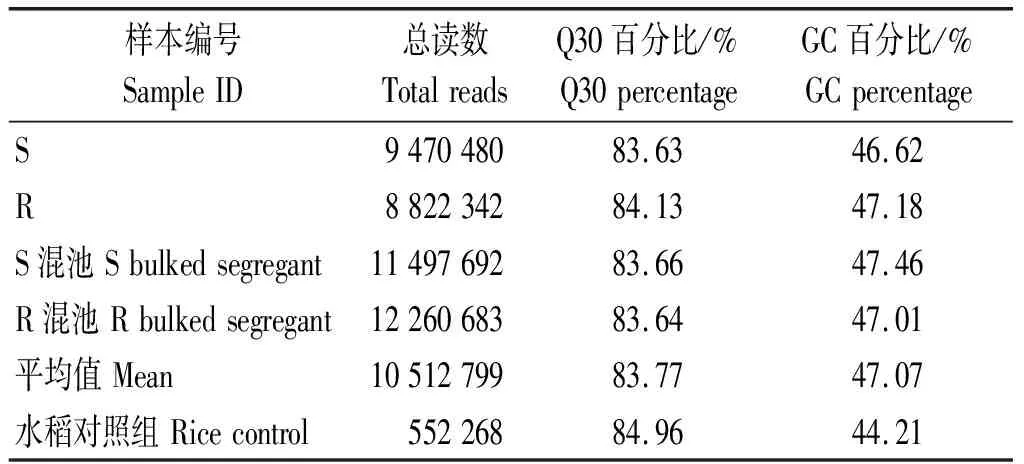
表2 各样品测序数据统计
2.3 SNP标记的获得及关联分析
利用玉米参考基因组共开发189 966个SLAF标签,供试样本中共开发获得696 139个SLAF标签,其中感病亲本(S)得到166 880个SLAF标签,抗病亲本(R)得到152 193个SLAF标签,S混池得到188 154个标签,R混池得到188 912个标签;供试亲本(S和R)的测序深度平均为40.30×,混池(S混池和R混池)测序深度平均为44.52×(表3)。根据测序结果读数在参考玉米基因组中的定位结果,通过GATK局部多重比对及Samtools变异检测2种方法得到变异位点的交集,获取SNP最终位点集,SNP信息见表4。根据玉米每条染色体中SLAF的分布情况,进一步绘制SLAF标签和SNP标签染色体分布图(图2)。

表3 SLAF标签统计
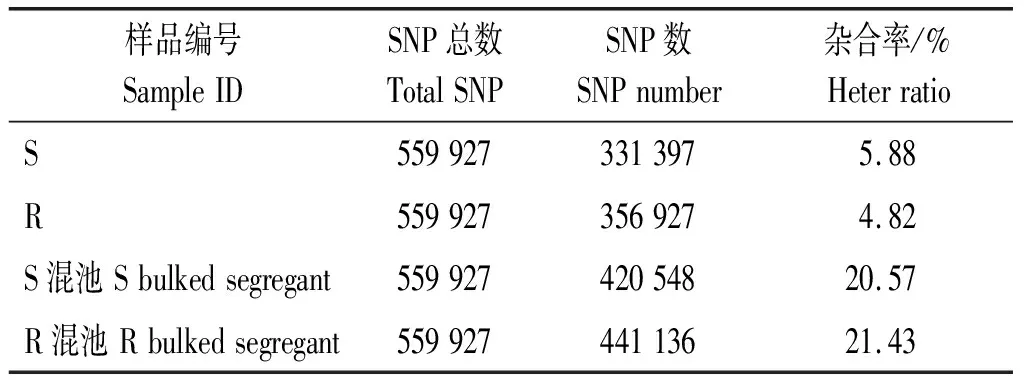
表4 SNP信息统计
图2 SLAF标签和SNP标记在染色体上的分布
采用SNP_index方法进行关联分析[8],首先过滤559 927个SNP位点,接着需过滤掉存在多重突变的408个SNP位点,然后再需过滤掉读数支持度<4的共计403 737个SNP位点,最后再过滤掉亲本中不存在的106 294个SNP位点,最终获取49 488个有效SNP位点。依据计算机软件系统模拟试验计算结果,当置信度为0.99时,定位起始区域位于第2号染色体14 002 645处,终止位置22 027 721处,关联区域大小为8.03 Mb,包含基因数量504个。使用Blast软件将关联区域内的504个基因分别在SwissProt、NR、KEGG、COG及GO 5个数据库中进行比对,最终获取376个基因的信息,并对关联区域内基因注释结果进行统计(表5)。结合已公布的玉米品种 B73 基因组序列,将目标基因定位区域基因组序列与 B73 全基因组序列进行比对,对目标基因初步定位所在区域的候选基因进行功能预测,分析亲本中外显子区域内具有差异的SNP位点,并进行变异注释(表6),在其中发现共有12个SNP位点存在非同义编码突变现象,可分别对应到6个基因上。进一步对候选区域内非同义突变对应的6个候选基因在GO、SwissProt、NR、COG及KEGG 5个数据库进行注释,基因注释信息见表7。由于NR、COG和KEGG 3个数据库中功能基因的有效注释信息过少,因此,表7仅列出GO和SwissProt 2个数据库的基因注释结果。对这6个候选基因的注释结果发现,这些基因的功能与丝氨酸/苏氨酸蛋白激酶活性、ATP及GTP结合、细胞信号转导及作为一些重要蛋白质的前体或结构域组成等方面相关。推测这些蛋白在监测病原菌入侵和随后的防御响应方面具有重要作用,作为与抗病性状直接相关的候选功能基因。今后还需进一步对这6个候选基因进行功能验证分析。
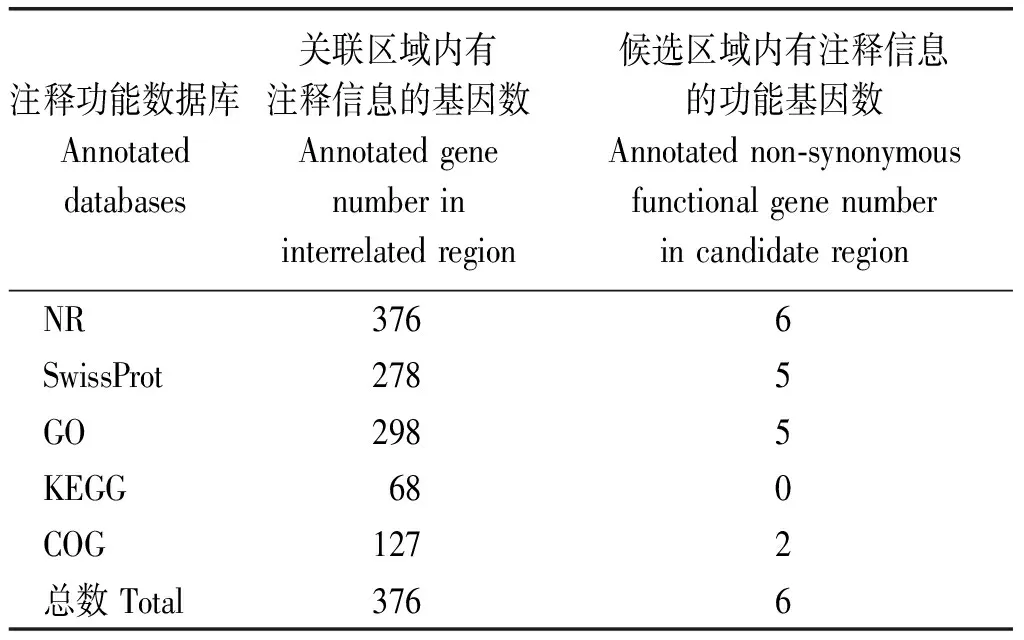
表5 关联区域及候选区域内非同义突变的基因功能注释结果统计

表6 SNP注释结果统计

表7 候选基因在GO和SwissProt数据库中的注释结果
2.4 关联区域内基因的GO富集分析
GO数据库作为一种标准生物学的结构化注释系统,该数据库建立了基因及其编码产物功能的相对分类体系,在生物物种中具有普适性。GO数据库的结构分成了多个层级,如层级越低则节点所代表的功能注释越详细。GO分析可按照细胞组成、分子功能和生物过程三大模块对所有注释的基因进行分类[9]。本研究关联区域内的基因在GO数据库中的分类统计结果如图3所示。以细胞组成模块中分析为例,在关联区域内基因的TopGO富集结果可以看出,所有基因注释到该部分的功能包括:胞外区、膜、细胞连接、大分子复合体、细胞器等;而在分子功能模块中,注释到该部分的基因功能包括核酸结合转录因子活性、催化活性、受体活性、鸟苷酸交换因子活性、运输活性、结合活性、电子载体活性、抗氧化剂活性、酶调节活性及分子传感器活性等。富集节点的显著性统计可以看出,其中GO:0033290、GO:0016282、GO:0005852富集显著性最高(表8)。
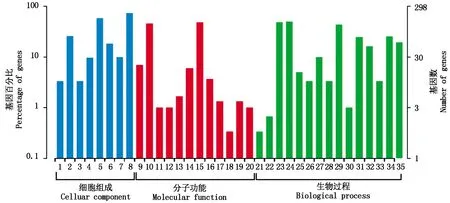
1.胞外区;2.膜;3.细胞连接;4.大分子复合体;5.细胞器;6.细胞器部分;7.膜部分;8.细胞部分;9.核酸结合转录因子活性;10.催化活性;11.受体活性;12.鸟苷酸交换因子活性;13.结构分子活性;14.运输活性;15.结合活性;16.电子载体活性;17.抗氧化剂活性;18.金属伴侣活性;19.酶调节活性;20.分子传感器活性;21.生殖;22.免疫系统代谢过程;23.新陈代谢过程;24.细胞过程;25.繁殖过程;26.多细胞生物过程;27.发育过程;28.生长过程;29.单细胞生物过程;30.生物阶段;31.应激反应;32.生物定位;33.多系统过程;34.生物调节;35.细胞成分组织或生物起源。
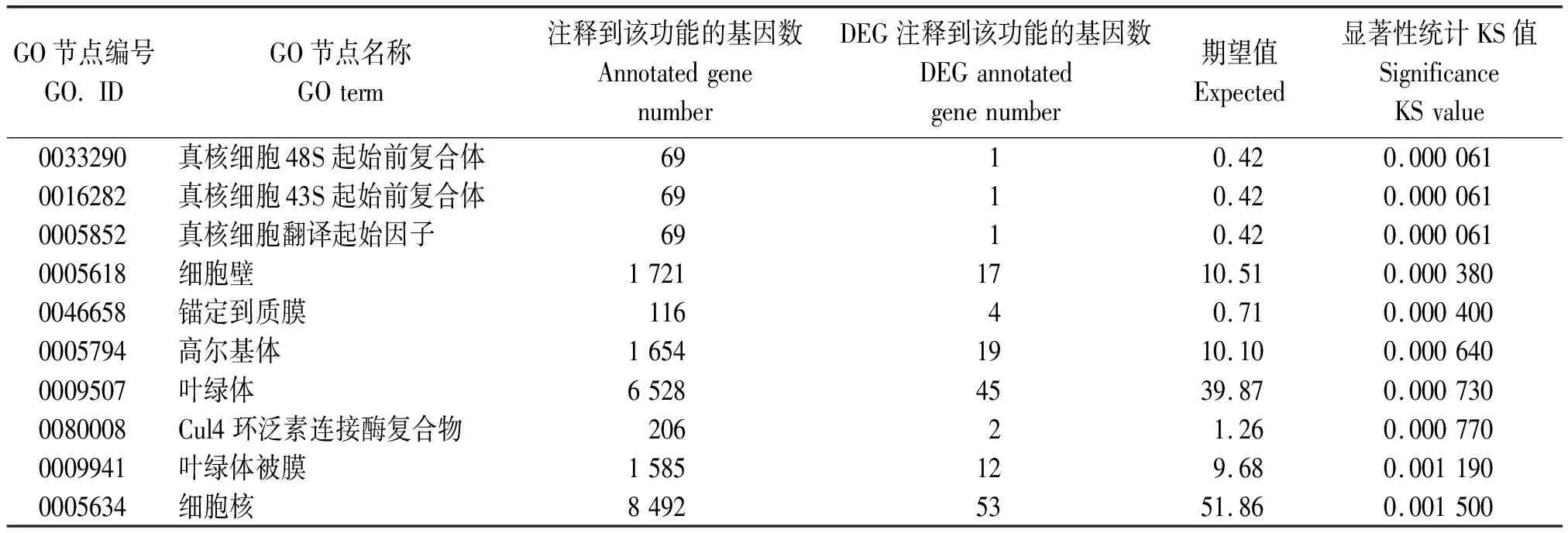
表8 关联区域内基因的TopGO富集结果(细胞组分)
3 讨论
本研究通过构建抗感病极端混池,利用SLAF-BSA技术对玉米尾孢菌抗灰斑病进行研究,将抗病相关基因关联区域定位于2号染色体 14 002 645~22 027 721区域内,获得6个与抗病相关的候选基因。宋军锋等[10]用BC2F4群体将抗病QTL定位在2号染色体,与本研究结果相符合,说明在2号染色体真实存在抗性QTL。此外,Bubeck等[11]通过RFLP分子标记技术对3个F2∶3群体进行了研究,结果在10条玉米染色体上均发现存在抗病QTL。Lehmensiek等[12]通过BSA法发现了11个与玉米灰斑病抗性相关的多态性AFLP标记,并将这些标记进一步转化为特异性PCR序列标记位点,发现其中5个标记分别与3个抗玉米灰斑病QTL存在连锁,分别定位在1,3,5号染色体上。Zhang等[13]利用高抗自交系Y32和高感自交系Q11组建的161个F2∶3家系群体进行玉米抗灰斑病QTL的初定位,扫描得到2个主效的QTL,qRgls1和qRgls2,分别位于bin5.04和bin8.02上。目前,针对玉米灰斑病抗性遗传研究主要以玉蜀黍尾孢菌侵染的灰斑病为主,对玉米尾孢菌仅限于病原学、发生规律的研究[2,14],已有研究结果并不能完全反映玉米尾孢菌灰斑病的抗病遗传规律。试验表明,引起灰斑病的2种病原菌的致病力有差异,玉米尾孢菌引起的玉米灰斑病,发病初期通常在叶片上形成退绿色的微型斑块,之后随着病情发展,病斑逐渐扩大形成淡灰色至浅褐色长条形斑,后期病斑联结成片状,病斑受叶脉限制,没有明显的边缘;而由玉蜀黍尾孢菌引起的玉米灰斑病在叶片边缘可见明显的褐色线形斑纹,形成的病斑在不同玉米品种上的大小及形状也可能存在差异,如遇到适宜的气候条件时,则病斑会加速蔓延扩展甚至引起整片叶子枯死。因此,应有针对性的加强抗玉米尾孢菌灰斑病抗性遗传机制研究,以有效控制该病害的流行与严重为害。
SLAF-seq技术作为一项高度自动化测序技术,以生物信息学、高通量测序等技术为基础,可获得全基因组分布的极大量的序列信息,高密度的序列信息可实现对候选功能区域的精细定位,高覆盖度和数字化信号为标签的准确性提供了保证,进而保证了基因定位准确性[15-16],已经运用在玉米、小麦、水稻等粮食作物上,并取得了显著的效果[17-19]。目前,SLAF和BSA技术已在高效开发利用植物基因组方面展开了应用,陈士强等[20]基于SLAF-seq技术,开发了20个长穗偃麦草1E染色体特异分子标记、2个长穗偃麦草基因组特异分子标记及26个其他特异性分子标记,基于优良性状与分子标记共分离的性质,最终获得抗性基因连锁相关的一系列分子标记。利用BSA已成功精细定位水稻真菌稻瘟病[21]、拟南芥抗寄生疫霉病基因位点[22]、小麦高籽粒蛋白含量基因GPC-B1[23]。SLAF-seq技术因其低成本、高特异性、高准确率、稳定可重复性好等优点,可大大缩短得到遍布于整个基因组海量序列信息的时间,有效地应用于标记开发,是实现候选功能区精细定位的一种有效的技术手段。
研究灰斑病抗病候选基因,发掘与抗病基因相连锁的分子标记,有助于研究植物的抗病机理,并为利用分子标记辅助改良抗性性状提供了理论支撑[14,24-25]。今后的工作重点是通过构建精细定位群体进一步对主效QTL进行定位和克隆,并在现有基础上搜寻与玉米灰斑病抗性基因紧密关联的分子标记,进而应用该标记对其衍生株系进行性状改良,或是将R225株系中的抗性基因导入至其他自交系,通过有利性状基因的聚合,从而研发更多抗灰斑病的优良玉米育种新材料。本研究结果为今后开展玉米尾孢灰斑病抗性QTL的精细定位和分子标记辅助育种奠定了一定的理论基础,对防控灰斑病的发生、减少该病对玉米生产的影响起到积极作用。
