Torin2 overcomes sorafenib resistance via suppressing mTORC2-AKT-BAD pathway in hepatocellular carcinoma cells
2021-01-07YiTingHuZheYueShuJingHuJingQinFenXieShuSenZheng
Yi-Ting Hu , Zhe-Yue Shu , Jing-Hu Jing , Qin-Fen Xie , Shu-Sen Zheng ,*
a Department of Hepatobiliary and Pancreatic Surgery, Shulan (Hangzhou) Hospital, Zhejiang Shuren University, Shulan International Medical College,Hangzhou 310022, China
b Division of Hepatobiliary Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310 0 03, China
c Department of Hepatobiliary and Pancreatic Surgery, Jinhua Municipal Central Hospital, Jinhua 3210 0 0, China
Keywords:Torin2 Sorafenib resistance Hepatocellular carcinoma mTORC2-AKT-BAD pathway
A B S T R A C T Background: Sorafenib is an oral multi-kinase inhibitor that was approved by the US Food and Drug Administration for the treatment of patients with advanced hepatocellular carcinoma (HCC). However, resistance to sorafenib is an urgent problem to be resolved to improve the therapeutic efficacy of sorafenib.As the activation of AKT/mTOR played a pivotal role in sorafenib resistance, we evaluated the effect of a dual mTOR complex 1/2 inhibitor Torin2 on overcoming the sorafenib resistance in HCC cells.Methods: The sorafenib-resistant Huh7 and Hep3B cell lines were established from their parental cell lines. The synergistic effect of sorafenib and Torin2 on these cells was measured by cell viability assay and quantified using the Chou-Talalay method. Apoptosis induced by the combination of sorafenib and Torin2 and the alteration in the specific signaling pathways of interest were detected by Western blotting.Results: Sorafenib treatment inversely inhibited AKT in parental but activated AKT in sorafenib-resistant Huh7 and Hep3B HCC cells, which underscores the significance of AKT activation. Torin2 and sorafenib synergistically suppressed the viability of sorafenib-resistant cells via apoptosis induction. Torin2 successfully suppressed the sorafenib-activated mTORC2-AKT axis, leading to the dephosphorylation of Ser136 in BAD protein, and increased the expression of total BAD, which contributed to the apoptosis in sorafenibresistant HCC cells.Conclusions: In this study, Torin2 and sorafenib showed synergistic cytostatic capacity in sorafenibresistant HCC cells, via the suppression of mTORC2-AKT-BAD pathway. Our results suggest a novel strategy of drug combination for overcoming sorafenib resistance in HCC.
Introduction
Globally, primary liver cancer is the seventh most common cancer and the second leading cause of cancer-related death. Among the primary liver malignancy, hepatocellular carcinoma (HCC) accounts for approximately 75% of the total cases [1] . HCC is natively resistant to traditional systemic chemotherapy [2] . Sorafenib is approved by US Food and Drug Administration in 2007 as the first-line drug for treatment of advanced HCC. However, the clinical efficacy of sorafenib was not satisfactory, as the median overall survival of patients that received sorafenib was only prolonged by 2-3 months compared with that of the placebo group in large-scale clinical trials [ 3 , 4 ]. Accumulating evidence showed that some patients with HCC responded to sorafenib in the initial stage but the tumor eventually progressed during sorafenib therapy [4] , indicating that the development of acquired resistance is a huge obstacle in sorafenib treatment.
The mechanisms involved in the sorafenib resistance in HCC are diverse. An increasing number of studies indicated that the compensatory effect and cross-talk between the signaling pathways,the generation of cancer stem cells, as well as the tumor microenvironment contributed to sorafenib resistance [ 5 , 6 ]. Since the therapeutic effect of sorafenib monotherapy is limited, the development of novel drug combination strategies is urgent to overcome the resistance to sorafenib.
Sorafenib is a multi-target kinase inhibitor, whose targets include receptor tyrosine kinases such as VEGFR2-3, PDGFR, FGFR-1 and c-Kit, as well as B-RAF and RAF-1 [7] . PI3K-AKT-mTOR and MAPK/ERK (RAS-RAF-MEK-ERK) signaling pathways are two important tumor-promoting downstream cascades of the above receptor tyrosine kinases. The MAPK/ERK pathway is frequently overactivated in HCC tissue [8] , which can be blocked directly by sorafenib through B-RAF and RAF-1 inhibition [9] . However, sorafenib does not directly suppress and even activates the PI3K/AKT/mTOR pathway, which confers resistance capacity of the HCC cells [10] .In a variety of pre-clinical studies, the efficiency of sorafenib combined with PI3K/AKT inhibitors for sorafenib-resistant HCC cells has been confirmed [ 11 , 12 ]. Nonetheless, the combination of dual mTOR complex 1/2 (mTORC1/2) inhibitor plus sorafenib still needs to be evaluated.
Torin2 is a novel second-generation ATP-competitive dual mTORC1/2 inhibitor [13] , which has shown the capacity of suppressing the proliferation in breast cancer, HCC, and ovarian cancer cells [14-16] . However, it remains unclear whether Torin2 may overcome the sorafenib resistance in HCC cell lines. Therefore, this study aimed to assess the cytostatic effect of Torin2 in combination with sorafenib for the treatment of sorafenib-resistant HCC cells and to explore the underlying mechanism.
Methods
Cell culture, chemicals and antibodies
Parental Hep3B human HCC cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Parental Huh7 human HCC cell line was purchased from Chinese Academy of Sciences Committee Type Culture Collection cell bank (Shanghai, China). HCC cell lines were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) (ATCC) plus 10% fetal bovine serum (HyClone, Marlborough, MA, USA). Cells were maintained in a humidified atmosphere of 5% CO 2 at 37 °C. The growth medium was replaced every other day and cells were passaged at 80% confluence. Sorafenib and Torin2 were purchased from Selleck Chemicals (Houston, Texas, USA). All antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA).
Establishment of sorafenib-resistant cell lines
The parental Huh7 and Hep3B HCC cells were exposed to 10μmol/L sorafenib for 24-48 h and then the residual cells were collected and transferred to a new flask. When the surviving HCC cells reached 75% -80% confluence, another round of the sorafenib exposure was carried out. After six months of continuous treatment,the sorafenib-resistant Huh7 cell line (Huh7-SR) and sorafenibresistant Hep3B cell line (Hep3B-SR) were established.
Cell viability and colony formation assays
The viability of HCC cells was measured by Cell Counting Kit 8(CCK-8) (Dojindo, Japan). Briefly, HCC cells were seeded into 96-well plates at 30 0 0 cells/well in 10 0μL growth medium for 24 h.Then cells were treated with sorafenib and/or Torin2 for 48 h. Before cell viability measurement, 10μL of CCK-8 reagent was added to each well followed by 4 h incubation. The plates were measured at OD450 nm with the BioTek Gen5 system (BioTeck, Winooski, VT,USA). For colony formation assay, the cells were seeded in six-well plates at 500 cells/well and cultured for 24 h. Next the cells were treated with sorafenib and/or Torin2 for 24 h, after which drugs were removed and the cells were cultured in drug-free medium for 14 days. Cell colonies were washed gently with PBS and fixed in 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet solution. The colonies containing>50 cells were counted.
IC 50 calculation and synergistic effect evaluation
The values of 50% inhibition concentration (IC50) for sorafenib were determined by Graphpad Prism 8 software (GraphPad Software, San Diego, CA, USA). The combination index (CI) value was determined from the fraction-affected value of each combination according to the Chou-Talalay method using CompuSyn software(ComboSyn, Inc., Paramus, NJ, USA) (CI = 1, additive effect; CI<1,synergistic effect; CI>1, antagonistic effect).
siRNA transfection
Transfections of siRNA (Tsingke Biotech, Beijing, China) were performed using LipofectamineR○30 0 0 (Thermo Fisher Scientific,Waltham, MA, USA) according to the manufacturer’s instruction.Briefly, cells were seeded into 6-well plates and reached 70%confluence at the time of transfection. siRNA-LipofectamineTM30 0 0 (Thermo Fisher Scientific) complexes in Opti-MEMTMreduced serum medium (Gibco, Waltham, MA, USA) were prepared and added to each well, and the final concentration of siRNA was 100 nmol/L. 24 h after transfection, the cells were ready for the next analysis.
Western blotting analysis
Cells were harvested using the RIPA cell lysis buffer and a total of 30μg proteins were loaded on 10% -12% SDS/PAGE gel. After electrophoresis the separated proteins were transferred onto PVDF membranes (Millipore, Billerica, MA, USA). After blocking,the PVDF membranes were incubated with 1:10 0 0 dilutions of specific primary antibodies overnight at 4 °C. Then the blots membranes were incubated with HRP-conjugated secondary antibodies at room temperature for 2 h and visualized using the BioSpectrum system (UVP, LLC, Upland, CA, USA). The protein band was quantified by ImageJ (Version 1.52a, NIH, Bethesda, MD, USA).
RNA extraction, reverse transcription, and quantitative real-time PCR analysis
Total RNAs were extracted using Trizol reagent (Invitrogen).2 μg of total RNA was reverse transcripted into cDNA using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific).The BAD (Hs00188930_m1) and GAPDH (Hs02786624_g1) primers were purchased from Thermo Fisher Scientific. Quantitative realtime PCR (qRT-PCR) was conducted using TaqMan PreAmp Master Mix Kit (Thermo Fisher Scientific) with ABI ViiA 7 System (Applied Biosystems, Waltham, MA, USA). The relative expression level of the target gene was normalized to the level of GAPDH mRNA using the 2-ΔΔCt method.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) and represented three or more independent experiments. All statistical tests were performed using the SPSS software (version 20.0, IBM Corp., Armonk, NY, USA). Differences between the two groups were calculated by unpaired Student’st-test. AP<0.05 was considered statistically significant.
Results
Confirmation of sorafenib resistance in Hep3B-SR and Huh7-SR cell lines

Fig. 1. Confirmation of sorafenib resistance in Huh7-SR and Hep3B-SR cell lines. Cells were treated with indicated concentration of sorafenib for 48 h.
A cell viability assay of parental and sorafenib-resistant Huh7 and Hep3B cell lines was carried out after the treatment of various concentrations of sorafenib ( Fig. 1 ). The IC 50 of sorafenib was 4.39-fold potent in the Huh7-SR (IC 50 = 13.42μmol/L) than that in the parental Huh7 (Huh7-P) (IC50= 3.05μmol/L) cells. Similarly, IC50of sorafenib was 3.66-fold potent in the Hep3B-SR (IC 50 = 13.52μmol/L) than that in the parental Hep3B (Hep3B-P) (IC 50 = 3.69μmol/L) cells, confirming that the in-house established Huh7-SR and Hep3B-SR cell lines were resistant to sorafenib treatment.
Sorafenib and Torin2 synergistically reversed sorafenib resistance
Monotherapy of Torin2 at the highest concentration of 50μmol/L showed modest cytostatic efficacy in sorafenib-resistant HCC cells, and the viability was reduced by 36% and 48.5% in Huh7-SR and Hep3B-SR cells, respectively ( Fig. 2 A). Though 2μmol/L of sorafenib slightly affected the viability of Huh7-SR and Hep3B-SR cells (Fig. S1A), and the addition of 2μmol/L of sorafenib significantly enhanced the potency of Torin2, as the inhibition curve was shifted down ( Fig. 2 A), indicating that sorafenib may act in synergy with Torin2. CI values were below one in most of the concentrations ( Fig. 2 B), indicating the significant synergistic inhibitory effects of sorafenib plus Torin2 for Huh7-SR and Hep3BSR cells. Similarly, parental cells were treated with 0.5μmol/L of sorafenib, a relative harmless concentration (Fig. S1B). The results showed that no significant synergistic effect exists in parental cells(Fig. S1C, D).
Moreover, the synergistic effect of sorafenib and Torin2 on sorafenib-resistant cell lines was verified by colony formation assay. We observed that sorafenib or Torin2 did not markedly abrogate the colony formation in Huh7-SR and Hep3B-SR cells, while the combination showed potent effectiveness on suppression of colony formation ( Fig. 2 C, D).
Sorafenib and Torin2 increased apoptosis in sorafenib-resistant HCC cells
Apoptosis induction is an important mechanism that mediates the anti-cancer effect of chemotherapy and targeted-therapy drugs.As demonstrated in Fig. 2 E, monotherapy of 10μmol/L sorafenib or 1μmol/L Torin2 induced moderate expression of cleaved-caspase3 and cleaved-PARP protein, which serve as markers of mitochondrial apoptosis, while the combination of these two drugs enhanced the cleavage of caspase3 and PARP in Huh7-SR and Hep3B-SR cells. Undoubtedly, apoptosis induction accounted, at least in part, for the synergistic action of sorafenib and Torin2.
Sorafenib activated mTORC2-AKT in Huh7-SR and Hep3B-SR cells but not parental cells
As shown in Fig 3 A, in Huh7-SR and Hep3B-SR cells, Ser473 site in AKT protein was phosphorylated by the treatment of sorafenib in a time-course manner, whereas, in parental Huh7 and Hep3B cells, AKT was dephosphorylated by sorafenib. Moreover, the phosphorylation level at Ser2481 was elevated after sorafenib treatment( Fig. 3 B), which was in line with the alteration of AKT. Additionally,we also observed the phosphorylation level of Ser473 in AKT and S2481 in mTOR, rather than the total protein level, was elevated in basal state in Huh7-SR and Hep3B-SR cells (Fig. S2). The result suggested that the mTORC2-AKT axis may play a pivotal role in sorafenib resistance in HCC cells and targeting mTORC2 might be a feasible way to overcome sorafenib resistance.
Inhibition of mTORC2 using Torin2 abrogated AKT-BAD axis
Compared to basal status, AKT was dephosphorylated in both Huh7-SR and Hep3B-SR cells (Fig. S3). Furthermore, as illustrated in Fig. 3 C, the phosphorylation of AKT was efficaciously reduced by Torin2 in the presence of sorafenib in both cell lines, indicating that AKT-inhibition might be a key factor for drugs combination treatment. Since Torin2 inhibits both mTORC1 and mTORC2,to exclude the possibility that mTORC1 may influence the activity of AKT, we treated Huh7-SR and Hep3B-SR cells with rapamycin,a well-known mTORC1 inhibitor, and a distinct different outcome was observed; rapamycin alone induced AKT phosphorylation and even enhanced this effect together with sorafenib ( Fig. 3 D). Therefore, we believe that Torin2 suppressed AKT via exerting its capacity of mTORC2 inhibition.
The basal phosphorylated BAD was increased in resistant cells,compared with that of parental cells (Fig. S2). Moreover, sorafenib induced BAD protein expression in resistant cells, and the combination of sorafenib and Torin2 further strengthened the expression of BAD ( Fig. 4 A). However, even if both the total and phosphorylated BAD were elevated after sorafenib treatment, the increase ratio was more significant in phosphorylated BAD ( Fig. 4 B), which means that the function of upregulated BAD was repressive. Torin2 effectively reversed the Ser163 phosphorylation of BAD, namely,Torin2 re-activated the pro-apoptotic function of BAD. Overall, our finding revealed that Torin2 abrogated the activity of AKT, followed by dephosphorylation of BAD. Thus, there are reasons to believe that the apoptosis induced by sorafenib plus Torin2 was BADmediated.

Fig. 2. Synergistic cytostatic effect of sorafenib combined with Torin2 in Huh7-SR and Hep3B-SR HCC cells. A: Dose-response in HCC cells to Torin2 with/without 2 μmol/L sorafenib treated for 48 h; B: Combination index of Huh7-SR and Hep3B-SR HCC cells treated with sorafenib and Torin2; C: Colony formation assay evaluating the proliferation of Huh7-SR and Hep3B-SR HCC cells treated with single agent or the combination of sorafenib (10 μmol/L) and Torin2 (1 μmol/L); D: Colony counting results of the colony formation assay; E: Sorafenib and Torin2 synergistically induced apoptosis in Huh7-SR and Hep3B-SR HCC cells detected by the expression of cleaved-capspase3 and cleaved-PARP using Western blotting. * P < 0.05, *** P < 0.001 compared to DMSO treatment group. FA: fraction affected.
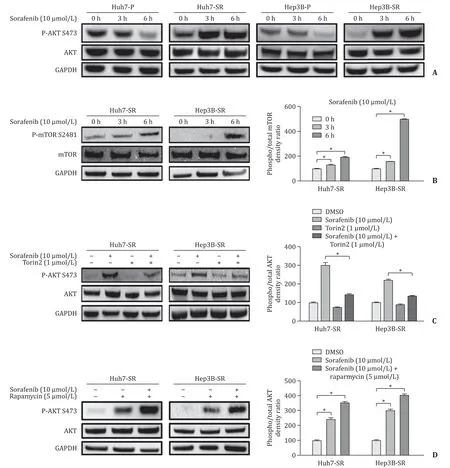
Fig. 3. Elevated activation of AKT induced by sorafenib was suppressed by Torin2 in sorafenib-resistant HCC cells. A: Western blotting results showing that sorafenib suppressed the phospho-AKT in parental HCC cells but increased the phospho-AKT in sorafenib-resistant cells; B: Sorafenib also increased phospho-mTOR in sorafenib-resistant cells. Torin2 reversed sorafenib induced AKT activation ( C ) while raparmycin did not ( D ). HCC cells were treated with indicated drug for 6 h before measurement. * P < 0.05.
BAD knockdown partially attenuated the cytostatic efficacy of sorafenib and Torin2 combination
After siRNA transfection, the mRNA as well as protein level of BAD was measured to confirm the knockdown efficiency (Fig.S4). The Huh7-SR and Hep3B-SR cells showed elevated cell viability and more resistance to the treatment of drug combination compared with negative control after BAD knockdown ( Fig. 5 A).Cleaved-caspase3 and cleaved-PARP were downregulated after BAD knockdown, especially in siR#1 and siR#2 groups ( Fig. 5 B).

Fig. 4. The expression of phosphorylated and total BAD after the treatment with single agent or the combination of sorafenib and Torin2 for 8 h was measured ( A ) and quantified ( B ). *** P < 0.001.

Fig. 5. BAD knockdown increased the resistance to the combination of sorafenib and Torin2 in HCC cells. A: Cell viability assay measuring the influence of BAD knockdown under the treatment of drug combination in Huh7-SR and Hep3B-SR cells; B: Apoptosis was reduced after BAD knockdown using siR#, as was detected by Western blotting.HCC cells were treated with 10 μmol/L sorafenib combined with 1 μmol/L Torin2 for 24 h. * P < 0.05, compared to siR-NC.
Discussion
Our study found that compared with monotherapy of each drug, the combined therapy of sorafenib and Torin2 suppressed the Huh7-SR and Hep3B-SR cell viability and the capacity of colony formation more effectively. Similarly, the pro-apoptotic effect in the sorafenib-resistant HCC cells was greater when the two drugs were used in combination.
AKT has been identified as a key factor that promotes sorafenib resistance in HCC cells and the phosphorylation of Ser473 is significant for its full activation [17] . In this study, we observed that sorafenib induced Ser473 phosphorylation in sorafenib-resistant cells rather than in parental cells. Additionally, in Huh7-SR and Hep3BSR cells, phosphorylation of Ser2481 in mTOR protein was elevated after sorafenib treatment. mTORC2 mainly consists of four subunits, including mTOR, mLST8, Rictor, and SIN1 [18] , and the phosphorylated Ser2481 in mTOR was proved to be a marker for the aggregation of mTORC2 complex [19] . Sorafenib suppresses RAF-1 and B-RAF, two proteins that mediate signal transduction in the MAPK/ERK signaling pathway [7] . Accumulating evidences have revealed that ERK inhibition generated by sorafenib suppresses mTORC1 [ 20 , 21 ], and mTORC1 was found to negatively regulate mTORC2 via IRS-1/PI3K in independent manner [22] . Therefore, the increased activity of mTORC2 by sorafenib exposure is logically sound. Given that AKT Ser473 is a mechanistic target of mTORC2 [23] , it is reasonable to infer that AKT phosphorylation was mTORC2-based. Furthermore, we tried to explore whether co-treatment of Torin2 and sorafenib may reverse the sorafenibinduced AKT phosphorylation in sorafenib-resistant HCC cells and it turned out that the combination successfully reduced the overactivation of AKT.
AKT was reported as a regulator of BAD that belongs to the BCL-2 family. BAD promotes the mitochondrial apoptosis by inhibiting other anti-apoptotic BCL-2 family members such as BCL-2 and BCL-XL [24] . In this study, we found that in sorafenib-resistant HCC cells, sorafenib alone increased BAD expression; nevertheless,the phosphorylated BAD was also increased and the rise of phosphorylated protein was even higher than the rise of total protein.Upon the combined treatment of sorafenib and Torin2, the phosphorylation level of BAD was reduced and the total BAD was further elevated. To further verify the speculation that upregulation of BAD induced apoptosis, BAD was knocked down in Huh7-SR and Hep3B-SR cells, which brought about increased resistance to the treatment of sorafenib combined with Torin2. Now that previous studies reported that BAD is phosphorylated by AKT at Ser136 and then switches into deactivated form [ 25 , 26 ], and the current results showed that the phosphorylation status of BAD was in line with the activation of AKT under the combination treatment, we have reasons to believe that the inhibition of AKT-BAD axis is the underlying mechanism that accounts for the synergistic cytostatic effect of these two drugs.
The PI3K/AKT/mTOR axis inhibition has been confirmed as a valid approach to overcome sorafenib resistance in HCC cells [ 11 , 12 ], whereas the specific mTORC2 inhibition was rarely reported in the field of sorafenib resistance. In this study, we discovered that not only AKT or PI3K/mTOR dual inhibition, but also mTORC2 inhibition is practicable to reverse sorafenib resistance.Nowadays, mTORC2 is emerging as an important driver in the process of tumorigenesis, cancer metabolic reprogramming [ 27 , 28 ]and targeted therapy resistance [29] . Thus, targeting mTORC2 is a promising choice for cancer treatment [30] . Recently, Werfel et al.reported that a nanoparticle-based RNAi mTORC2 inhibition improved the effectiveness of lapatinib on the treatment of breast cancer [31] . However, it is regrettable that no mTORC2 specific small-molecule inhibitor was available. Best to our knowledge, the present study is the first to report that the dual mTORC1/2 inhibitor Torin2 works synergistically with sorafenib to overcome sorafenib resistance in HCC cells.
There were some limitations in our study. First, this study was conductedinvitro,andinvivoexperiments need to be performed to verify the current conclusion. Besides, Torin2 is only used in pre-clinical studies, which holds a long distance away from the clinical trial. Nonetheless, we believe that all the findings are promising clues for Torin2 or even mTORC2 inhibition in the treatment of sorafenib-resistant HCC. Our future research will focus oninvivostudy to validate current conclusions.
Acknowledgments
None.
CRediT authorship contribution statement
Yi-Ting Hu:Investigation, Methodology, Writing - original draft.Zhe-Yue Shu:Methodology, Writing - original draft.Jing-Hua Jiang:Methodology, Software.Qin-Fen Xie:Data curation, Funding acquisition.Shu-Sen Zheng:Conceptualization, Supervision, Writing - review & editing.
Funding
This study was supported by a grant from Medical and Health Science and Technology Program of Zhejiang Province (2019RC076).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.hbpd.2020.09.010 .
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Non-operative management of pancreatic trauma in adults
- Hepatocellular carcinoma incidence post direct-acting antivirals in hepatitis C-related advanced fibrosis/cirrhosis patients in Australia
- Efficacy and cost-effectiveness of antiviral regimens for entecavir-resistant hepatitis B: A systematic review and network meta-analysis
- Combined hepatocellular-cholangiocarcinoma: An update on epidemiology, classification, diagnosis and management
- Hepatobiliary&Pancreatic Diseases International
- Safety and efficacy of an integrated endovascular treatment strategy for early hepatic artery occlusion after liver transplantation