清瘟败毒颗粒中连翘苷的薄层色谱鉴别和含量测定试验
2021-01-06赵玉丛李艳玲郑瑞锦张远方
赵玉丛, 李艳玲, 郑瑞锦, 张远方
(河南牧业经济学院制药工程学院, 河南郑州450046)
清瘟败毒散是《中华人民共和国兽药典》(简称《中国兽药典》)收载的成方制剂[1],具有泻火解毒、清热凉血的功效,主治畜禽热毒发斑、高热神昏等,临床上主要用于治疗禽流行性感冒、传染性胸膜肺炎、急性腹膜炎、病毒性腹泻及不明原因的顽固性水泻、肠毒综合征、各种细菌及原虫感染等引起的畜禽疑难杂症等[2-5]。
清瘟败毒颗粒是清瘟败毒散的改剂型制剂,将清瘟败毒散制成现代制剂颗粒剂,可克服散剂在临床使用中吸收利用率低、见效慢、单位体重用量大等不足,同时在制备颗粒剂时所用的辅料蔗糖会起到矫味剂的作用,增加其适口性,有利于猪和其他大动物的临床使用。目前《中国兽药典》所收载的清瘟败毒散的质量标准仅有显微鉴别和黄连的薄层色谱(TLC)鉴别,无含量测定项,但散剂可用显微鉴别各成分,颗粒剂由于不具备散剂的原始纤维或药材结构特点,故不能使用显微鉴别。《中国兽药典》目前亦尚未收载清瘟败毒颗粒。本课题组拟通过对清瘟败毒颗粒中的指标成分进行TLC鉴别和含量测定的方式以建立其质量标准。连翘是清瘟败毒颗粒中的重要药味,其主要药效成分连翘苷具有抗菌、抗病毒、强心、抗肝损伤等作用。连翘苷的含量测定方法文献报道较多,有近红外光谱(FT-NIR)法[6]、高效液相色谱(HPLC)法[7-9]、高效液相色谱-质谱联用(HPLC-MS)法[10]、超高效液相色谱(UPLC)法[11]等,其中HPLC法获得了广泛的应用,但尚未见用HPLC法测定清瘟败毒颗粒中连翘苷的含量的报道。本试验选择连翘苷作为清瘟败毒颗粒剂的指标成分之一,建立了连翘苷的TLC鉴别方法和HPLC含量测定方法,并对连翘苷从连翘药材到清瘟败毒散、连翘药材到清瘟败毒颗粒中的转移率进行对比研究,以期为清瘟败毒颗粒剂制备工艺的优化和质量标准的建立提供依据。
1 材料
LabTech-LC18型液相色谱仪(北京莱伯泰科仪器有限公司) ; KQ-200KDE超声波清洗机(昆山市超声仪器有限公司) ; SHZ-96A型循环水式真空泵(巩义市华予仪器有限责任公司) ; 分析天平(上海医用激光仪器厂) ; AL-100A型多功能摇摆式粉碎机(上海宾润电器有限责任公司)。
乙腈、甲醇均为色谱纯(上海金太阳化学品有限公司) ; 冰醋酸、石油醚(30~60 ℃)、三氯甲烷等其他试剂均为分析纯(湖北杜克化学科技有限公司)。连翘苷对照品,购自中国药品生物制品检定所; 清瘟败毒颗粒和阴性样品,河南牧业经济学院制药工程学院提供,阴性样品不加连翘药材 ; 连翘药材,购自河南郑州瑞龙国药医药有限公司,经河南牧业经济学院制药工程学院赵建平高级兽医师检验,符合2015版《中国兽药典》(二部) 项下相关要求和规定。取连翘药材约100 g,粉碎过5号筛。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液的制备 精密称定连翘苷对照品20.000 mg于10 mL容量瓶中,加甲醇稀释至刻度,制成每1 mL含2 mg的溶液,作为对照品贮备液。分别精密量取此贮备液适量,置于10 mL容量瓶中,加甲醇定容至刻度,摇匀,制成浓度分别为0.025、0.05、0.1、0.2、0.4 mg/mL和0.8 mg/mL的对照品溶液。
2.1.2 供试品溶液的制备 取清瘟败毒颗粒样品15 g,精密称定,置具塞锥形瓶中,加石油醚(30~60 ℃)20 mL密塞,超声15 min,滤过,弃去石油醚液,加甲醇20 mL密塞,超声20 min,滤过,滤液浓缩至1 mL,作为供试品溶液。
2.1.3 阴性对照液的制备 取缺连翘的清瘟败毒颗粒(制药工程学院自制)15 g,精密称定,按供试品溶液的制备方法得阴性对照液。
2.1.4 连翘药材对照溶液的制备 取连翘药材粉末1 g精密称定,按“2.1.2”项下方法,制得连翘药材对照溶液。
2.2 连翘苷的TLC鉴别 吸取连翘阴性对照液、清瘟败毒颗粒供试品溶液、连翘苷对照品溶液和连翘药材对照液各3 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇(8∶1)为展开剂,展开,取出,晾干,喷10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,检视,结果见中插彩版图1。由图1可知,供试品色谱中在与连翘对照药材色谱和连翘苷对照品色谱相应的位置上,显相同颜色的斑点,而阴性色谱没有,表明本试验建立的TLC法可以进行清瘟败毒颗粒中连翘苷成分的定性鉴别。
2.3 连翘苷的HPLC含量测定
2.3.1 色谱条件 色谱柱:LabTech-C18(4.6 mm×150 mm,5 μm);流动相∶水∶冰醋酸(27∶73∶0.1);流速:1.0 mL/min;UA检测波长:277 mm;柱温:30 ℃; 进样量:10 μL。
2.3.2 专属性试验 分别精密吸取上述对照品溶液、供试品溶液、阴性对照液和连翘药材对照溶液各10 μL,在2.3.1项色谱条件下进样测定,记录色谱图,结果见图2、3、4、5。结果显示,供试品中连翘苷与对照品色谱峰保留时间一致(保留时间为14.638 min),与其他共存成分的分离度良好(>1.5),且阴性样品在相应位置无色谱峰干扰,表明清瘟败毒颗粒中其他成分对连翘苷的测定无干扰。
2.3.3 线性关系考察 精密吸取各浓度连翘苷对照品溶液10 μL分别进样,按2.3.1项色谱条件测定,记录峰面积(A)为纵坐标,以连翘苷浓度(C)为横坐标,进行线性回归,得连翘苷的回归方程:A=6 810 842.36C+17 742.88(r=0.999 9)。线性结果表明,连翘苷的浓度在0.025~0.8 mg/mL范围内呈良好的线性关系。
2.3.4 精密度试验 通过测定连翘苷对照品的日内、日间精密度可以进行精密度的评价,分别选取连翘苷对照品高、中、低3个不同浓度(分别为0.4、0.2 mg/mL和0.05 mg/mL),日内精密度的考查为在同一天内每个浓度重复测定6次,日间精密度则是在连续3 d每天每个浓度重复测定2次,并计算相对标准偏差(RSD),评价方法的重现性。结果显示,连翘苷的日内精密度RSD和日间精密度RSD分别小于0.9%和1.8%,表明仪器精密度良好。
2.3.5 回收率试验 精密称取清瘟败毒颗粒9份,每份15 g,分别加入高、中、低3个浓度的连翘苷对照品溶液,每个浓度3份,按2.1.2项方法制备溶液,在2.3.1项色谱条件下分别进样10 μL进行测定,计算回收率和RSD(见表1),得连翘苷的平均回收率在96.4%~103.2%,RSD范围为1.0%~4.5%。

图2 连翘苷对照品色谱图Fig. 2 Chromatogram of Forsythin reference substance 1:连翘苷; 下图同1:Forsythin. The same as below figures

图3 供试品色谱图Fig. 3 Chromatogram of test sample

图4 阴性对照色谱图Fig. 4 Chromatogram of negative control

图5 连翘药材对照色谱图Fig. 5 Control chromatogram of Forsythia suspense
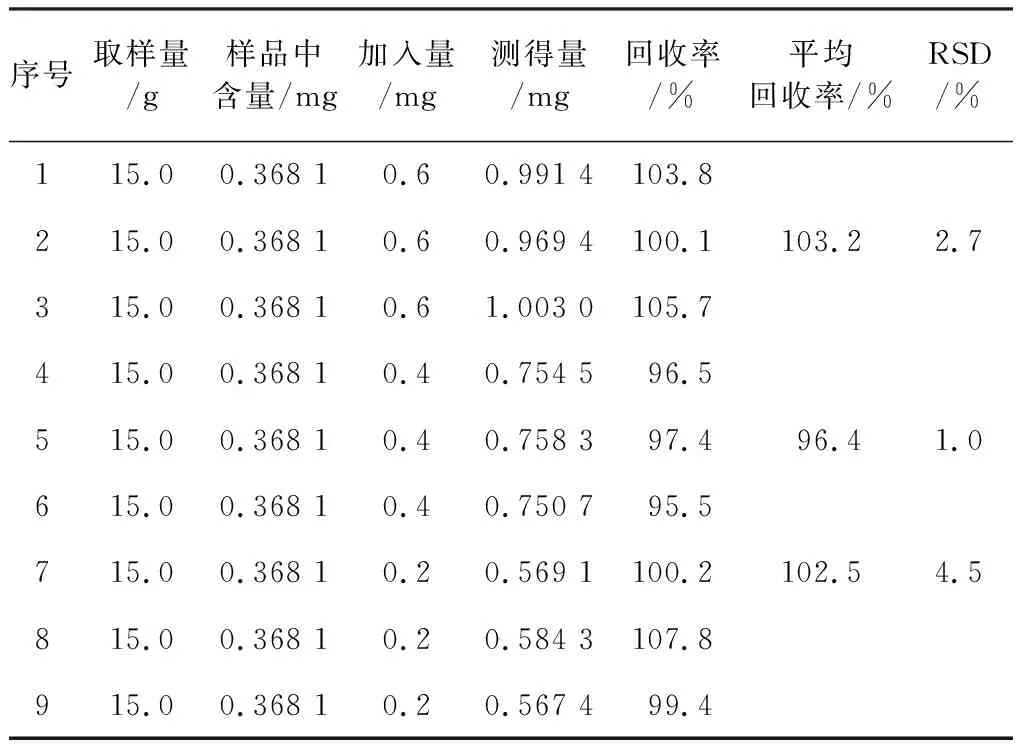
表1 回收率试验结果Table 1 Recovery test results
2.3.6 样品含量测定 分别取连翘药材以及由同一批药材制得的清瘟败毒颗粒样品和清瘟败毒散样品(均为自制,15 g颗粒剂和散剂分别相当于1 g连翘药材)各3 批,按2.1.2项方法制备样品溶液,在2.3.1项色谱条件下依次测定,结果见表2。从表中数据可以看出,连翘苷在连翘药材中的含量为1.526 5 mg/g,在清瘟败毒颗粒样品中的含量为0.720 1 mg/15 g,连翘苷从药材到颗粒剂的转移率为47.2%;连翘苷在清瘟败毒散样品中的含量为0.721 9 mg/15 g,连翘苷从药材到散剂的转移率为47.3%。两者之间的转移率并无明显差异。

表2 样品测定结果Table 2 Sample determination results (n=3)
3 讨论
3.1 方法的选择 本试验分别选择了TLC法和HPLC法进行清瘟败毒颗粒中连翘苷的鉴别和含量测定,结果表明,本试验所建立的TLC鉴别方法专属性强,HPLC含量测定方法在专属性强的前提下,精密度及线性关系较好,回收率亦较为满意,可用于清瘟败毒颗粒中指标成分连翘苷的质量控制。
3.2 流动相的选择 根据文献报道,HPLC法测定制剂中连翘苷含量的方法主要采用乙腈-水流动相系统[7-9],本试验参照以上方法多次调整溶剂系统中乙腈和水的比例,最后确定乙腈-水比例为27∶73,该比例下连翘苷色谱峰保留时间14.638 min,与制剂中其他成分的色谱峰分离良好,鉴于色谱峰出现拖尾现象,又添加适量的冰醋酸调整流动相的pH,使其峰形有所改善,最终确定流动相为乙腈∶水∶冰醋酸(27∶73∶0.1)系统。
3.3 连翘苷提取工艺的优化 在2015版《中华人民共和国药典》中,连翘苷的提取需经过氧化铝柱[7],但中性氧化铝在去除杂质的同时,连翘苷也因吸附而造成损失,本试验在保证连翘苷与制剂中其他成分完全分离的基础上,选择不过氧化铝柱,可减少连翘苷提取过程中的损失,精简提取步骤,提高分析效率;提取溶剂的考察,比较了水、乙酸乙酯、甲醇等不同溶剂提取后得到的色谱峰峰面积,确定甲醇为最佳提取溶剂;提取方式比较了超声提取、加热回流和静置过夜这3种常用的提取方法,试验结果显示,超声提取法优于其他2种方法;超声提取时间比较了超声处理10、20、30 min和40 min的差别,结果表明,超声提取20 min后,连翘苷的含量基本恒定。本试验最终确定连翘苷的提取工艺为先用石油醚脱脂,然后用甲醇超声提取20 min,该方法制备得到的供试品可同时满足连翘苷的TLC鉴别和HPLC含量测定。
本试验结果表明,由同一批药材制得的颗粒剂与散剂中连翘苷的含量无明显差别,说明单就指标成分连翘苷来看,从化学成分上清瘟败毒颗粒替代传统散剂应用可行。但中药制剂的质量不能单以一个或几个指标成分来进行评价,研究清瘟败毒颗粒在临床上替代传统散剂的可行性,还需结合药理药效、药代动力学参数和临床效果等进一步的研究。
连翘苷从药材到散剂及从药材到颗粒剂的转移率均偏低,一方面是因为散剂一般为粗粉直接入药,细胞破壁率极低,同时因为药物粒粗,位于粒子内部的有效成分需穿过几个或数十个细胞壁及细胞膜方可释放出来,造成释放速度很慢;而连翘苷从药材到颗粒剂的转移率低,表明清瘟败毒颗粒剂的生产工艺需进一步改进。
