基于生物信息学的宫颈癌枢纽基因筛选及其与预后的关系*
2021-01-05许修颖龚荣府杨娉娉方文
许修颖,龚荣府,杨娉娉,方文
(贵州医科大学 医学检验学院 生化教研室,贵州 贵阳 550004)
据2018年世界卫生组织统计,宫颈癌(cervical cancer,CC)是全球女性癌症发病率和死亡率均位居第4位的癌症[1]。随着医疗技术的提高、CC筛查的普及、生活方式的改变,CC发病呈年轻化趋势[2],研究发现CC仍然是20~39岁女性癌症死亡的第二大原因[3]。在欠发达国家,CC的发病率及死亡率过高,可能是因为筛查机会减少和人类乳头状瘤病毒(humanpapillomavirus,HPV)疫苗的高成本[4]。因此,仍有必要寻找与CC早期诊断、治疗和预后密切相关的靶点基因。近年来,越来越多的研究人员将基因图谱和基因芯片应用于科学研究[5],认为大多数基因芯片或基因图谱数据只存储在数据库中未被充分利用,重新分析这些数据可为研究癌症提供新的方法[6],研究人员利用生物信息学方法分析肝细胞癌、肺癌及乳腺癌等多种癌症的治疗靶点[7-9]。近期研究中,有学者通过分析基因表达数据库(gene expression omnibus,GEO)中GSE103512数据集,筛选出在宫颈癌组织中表达水平明显增加的基因,并认为是宫颈癌治疗的靶标[10]。然而,只从一个数据集中筛选表达升高的基因作为治疗的靶标的研究并不全面,本研究通过分析GEO中CC的多个基因表达数据集,采用多种生物信息学方法筛选调控CC的枢纽基因,并利用肿瘤基因组图谱(the cancer genome atlas,TCGA)验证枢纽基因的表达。
1 资料与方法
1.1 资料
从GEO(https://www.ncbi.nlm.nih.gov/geo/)中筛选CC微阵列数据集,输入关键字“cervical cancer”,选择“series”、“home sapiens”、“expression profiling by array”,最后得到134个“series”。通过阅读摘要,本研究选择了GSE9750、GSE7083和GSE63514作为数据来源,在基因表达谱中选择正常宫颈(normal cervix,NC)组织、高级别鳞状上皮内病变(high grade squamous intraepithelial lesion of the cervix,HSIL)、宫颈上皮内瘤样病变(cervical intraepithelial neoplasia,CIN)及CC组织样本进行后续分析。见表l。

表1 CC相关数据信息Tab.1 Data information on CC
1.2 方法
1.2.1筛选差异基因 在R(vesion:3.6.1)语言环境下,R-Studio利用GEOquery、limma、ggplot2等软件包处理3个数据集,根据表1中选择样本分组后筛选出差异表达基因。定义差异基因的筛选标准如下:P<0.05,且|log2FC|>1。获得差异基因后,用火山图展现3个数据集的差异基因,取3个数据集的差异基因交集,并用VennDiagram软件包绘制韦恩图。
1.2.2差异基因的富集分析 使用database for annotation、visualization and integrated discovery网站(DAVID,Vision:6.7,https://david-d.ncifcrf.gov/)阐明相互作用基因的生物学过程和信号通路[11- 12]。通过DAVID在线分析的方式获得差异基因在基因本体(gene ontology,GO)与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路分析中具体的富集情况,其中GO分析分为生物学过程、细胞定位CC及分子功能。
1.2.3蛋白质相互作用(protein-protein Interactions,PPI)网络分析及筛选枢纽基因 将差异基因导入STRING数据库来绘制相互作用基因并构建PPI网络[13],将PPI网络数据导入Cytoscape软件(Vision:3.7.2),利用Cytoscape插件CytoHubba预测和查找网络中的重要节点和子网络,采用12种拓扑分析方法,包括degree、edge percolated component(EPC)、maximum neighborhood component (MNC)、density of maximum neighborhood component (DMNC)等[14],从每种方法中选择前10个基因,计算所有选择出的基因在12种算法中的出现次数,最终选取次数最多的前10个基因作为枢纽基因,最终所有鉴定的枢纽基因被用来构建一个完整的PPI网络。
1.2.4枢纽基因的筛选及表达 采用 The Kaplan Meier plotter (http://kmplot.com/analysis/)网站在线分析枢纽基因[15],数据来自于TCGA数据库的宫颈癌(CC:n=304)临床信息,筛选与CC总体生存率(overall survival,OS)相关的基因。UALCAN(http://ualcan.path.uab.edu/)是一个基于 TCGA 数据集,肿瘤基因表达分析的在线数据库[16],本研究在UALCAN网站下载TCGA宫颈癌相关数据NC(n=3)和CC(n=305),并分析枢纽基因的表达情况。
1.2.5人类蛋白质图谱(the human protein atlas,HPA)数据库的分析 从HPA[17]中分别调取CC和NC组织中的细胞周期蛋白依赖性激酶1(cyclin dependent kinase 1,CDK1)、驱动蛋白家族成员11(kinesin family member 11,KIF11)、细胞周期蛋白B1 (cyclin B1,CCNB1)、细胞分裂周期蛋白45 (cell division cycle 45,CDC45)及CXC趋化因子配体8(c-x-c motif chemokine ligand 8,CXCL8)蛋白的免疫组织化学结果,明确其在CC和NC组织中的蛋白表达。
2 结果
2.1 差异基因的筛选
经R-Studio软件分析,分别从数据集GSE7083、GSE9750及GSE63514中得到差异表达基因760、1 454及3 019个,其中上调差异基因分别为393、785及1 880个,下调差异基因分别为367、669及1 139个。分别用火山图显示3个数据集差异基因(图1),全面系统地剖析3个数据集中的差异表达基因,经过VennDiagram软件包交集3个数据集差异基因后获得401个共同差异表达基因,包括219个上调基因和182个下调基因(图2)。
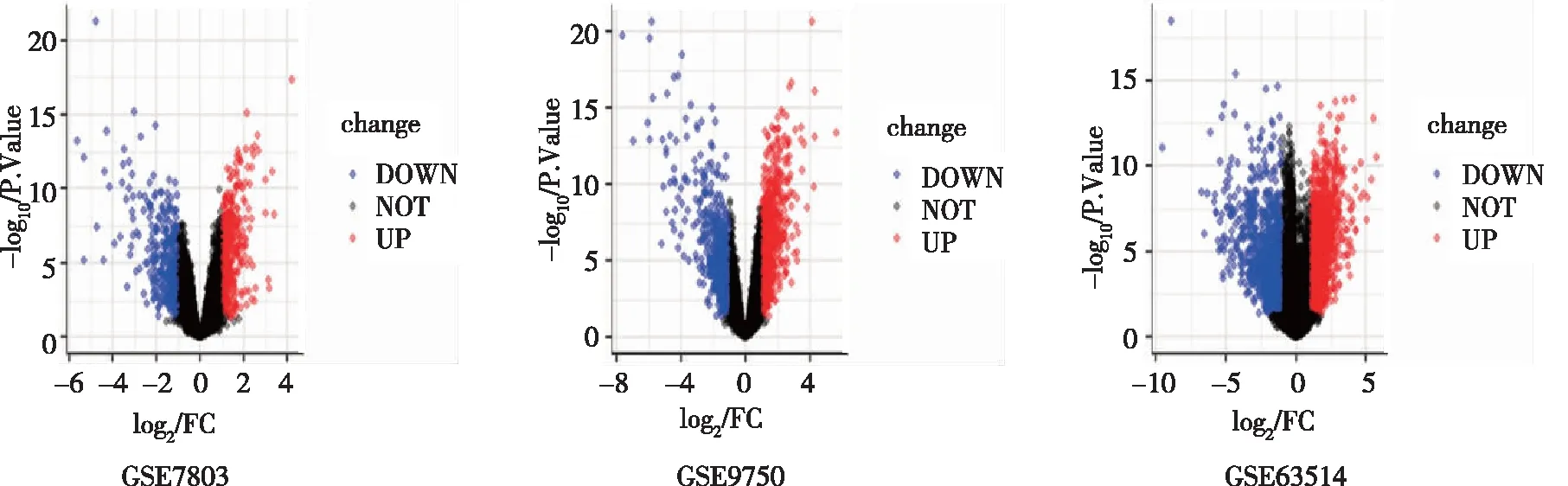
注:A为GSE7803,上调差异基因个数为393,下调差异基因个数为367;B为GSE9750,上调差异基因个数为785,下调差异基因个数为669;C为GSE63514,上调差异基因个数为1 880,下调差异基因个数为1 139;蓝色为下调差异基因,红色为上调差异基因,黑色为无显著差异基因。图1 GSE7803、GSE9750及GSE63514差异表达基因的火山图Fig.1 Volcano plot of GSE7803,GSE 9750, and GSE 63514 DEGs
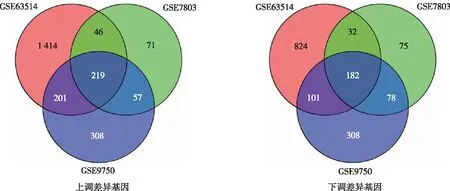
注:绿色、蓝色及红色圆圈分别为GSE7803、GSE9750、GSE63514差异基因个数。图2 上调和下调差异基因交集的韦恩图 Fig.2 Venn diagram of up-regulation and down-regulation of intersection of DEGs
2.2 差异基因的富集分析
GO分析结果显示,DEGs的细胞定位主要在染色体、无膜细胞器、角质包膜及纺锤体中。在生物学过程的分析中,DEGs与细胞周期、有丝分裂细胞周期、细胞周期过程及M期有关;DEGs的分子功能主要富集在丝氨酸型内肽酶活性、丝氨酸型肽酶活性、丝氨酸水解酶活性、细胞周期素依赖性蛋白激酶调节活性及内肽酶活性(表2)。DEGs的KEGG通路富集结果显示,细胞周期、DNA复制、卵母细胞减数分裂、p53信号通路及花生四烯酸代谢是主要途径(表3)。

表2 差异基因的GO富集分析Tab.2 GO enrichment results of DEGs

表3 差异基因的KEGG通路分析Tab.3 KEGG pathway analysis of DEGs
2.3 枢纽基因的筛选及PPI网络构建
401个DEGs被用于构建PPI网络,结果表明,PPI网络具有明显高于预期的交互作用(P<1.0×10-16),节点数为398,边缘数为4 899。经过12种算法的计算,由于第11个基因与第10个基因在12种算法中出现次数相同,故本研究共筛选出 11个枢纽基因(CDK1、KIF11、BUB1B、CCNB1、CCND1、CDC20、CDC45、CXCL8、ECT2、ESR1及TOP2A,表4),其中除了ESR1和CCND1基因在CC组织中表达下调外,其余基因表达上调;11个枢纽基因重新导入STRING数据库后构建PPI网络显示蛋白间有较高的交互作用(P=1.95×10-10,图3)。
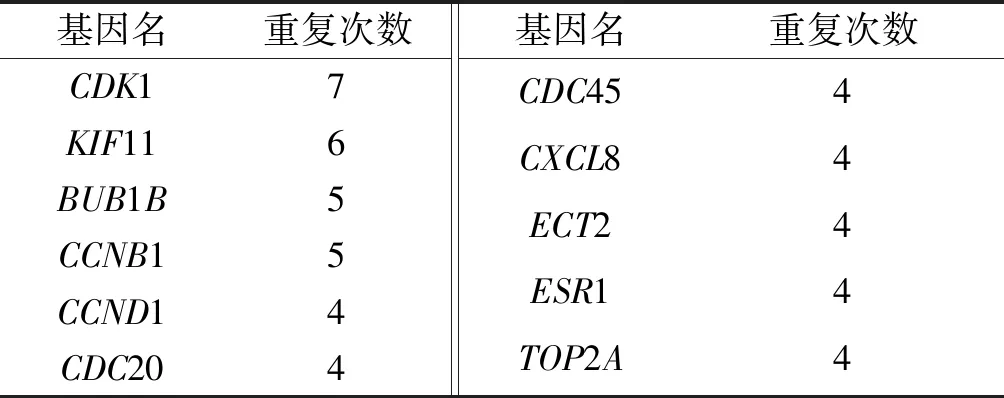
表4 枢纽基因在12种算法中的出现次数Tab.4 Number of occurrences of hub genes in 12 algorithms
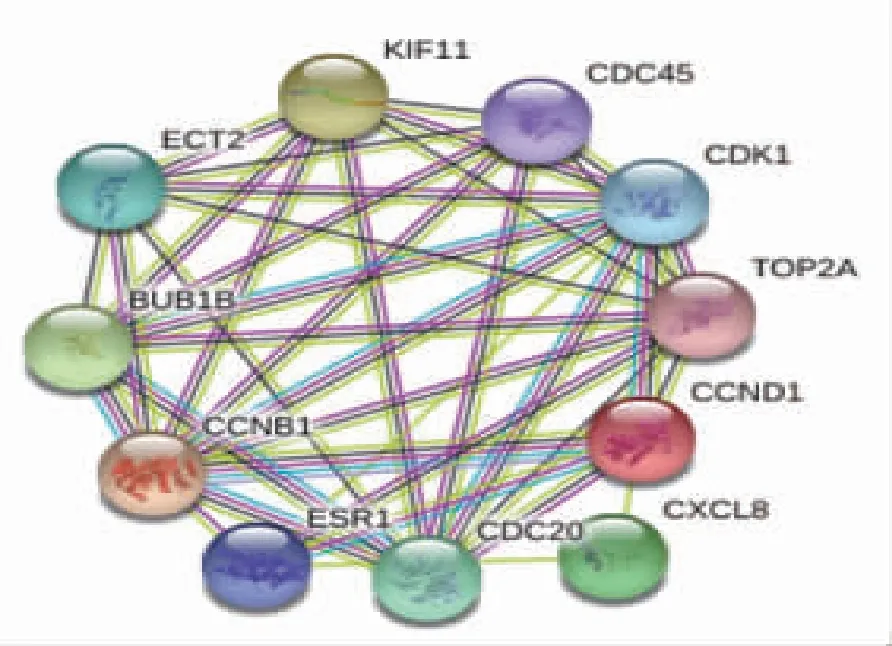
图3 枢纽基因的PPI网络Fig.3 PPI networks of hub genes
2.4 枢纽基因的生存分析及TCGA数据库验证枢纽基因的表达结果
在The Kaplan Meier plotter网站中收录304例CC患者数据中,CDK1、KIF11、CCNB1、CDC45及CXCL8基因的表达水平对患者的总生存时间有着显著影响;与低表达组相比,CDK1、KIF11、CCNB1及CDC45高表达组的CC患者的总生存时间增高(P<0.05);与低表达组相比,CXCL8高表达组的CC患者的总生存时间明显降低(P<0.001,图4)。UALCAN分析结果表明,CDK1、KIF11、CCNB1、CDC45及CXCL8基因在CC组患者中表达较NC组明显上调(P<0.01,图5)。
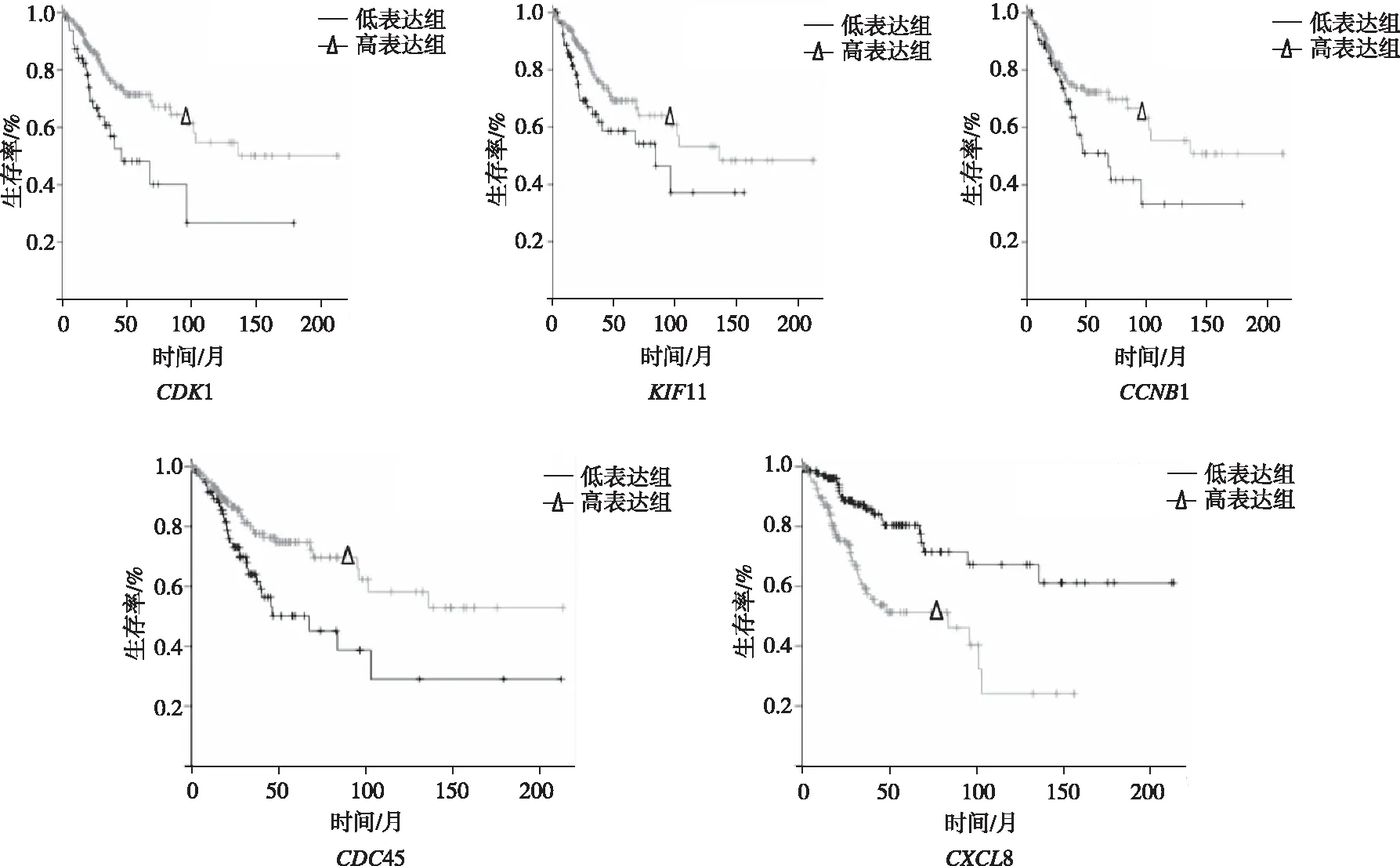
图4 枢纽基因高、低表达组CC患者预后的Kaplan-Meier分析Fig.4 Kaplan-Meier analysis of overall survival in CC patients with hub genes high and low

注:(1)与NC组比较,P<0.01。图5 CC组与NC组枢纽基因的表达Fig.5 The expression of hub genes in CC and NC groups
2.5 HPA数据库分析
HPA数据库中,采用不同的免疫组织化学抗体分析NC组织和CC组织的免疫组化结果及5种蛋白在CC组肿瘤细胞及对照组宫颈细胞中的定位(表5),CDK1、KIF11、CCNB1及CDC45蛋白相对于NC细胞在CC肿瘤细胞中表达增加,但CXCL8蛋白在CC及NC组织中都未检测到(图6),证实CDK1、KIF11、CCNB1及CDC45蛋白在CC组中较NC组织高表达。
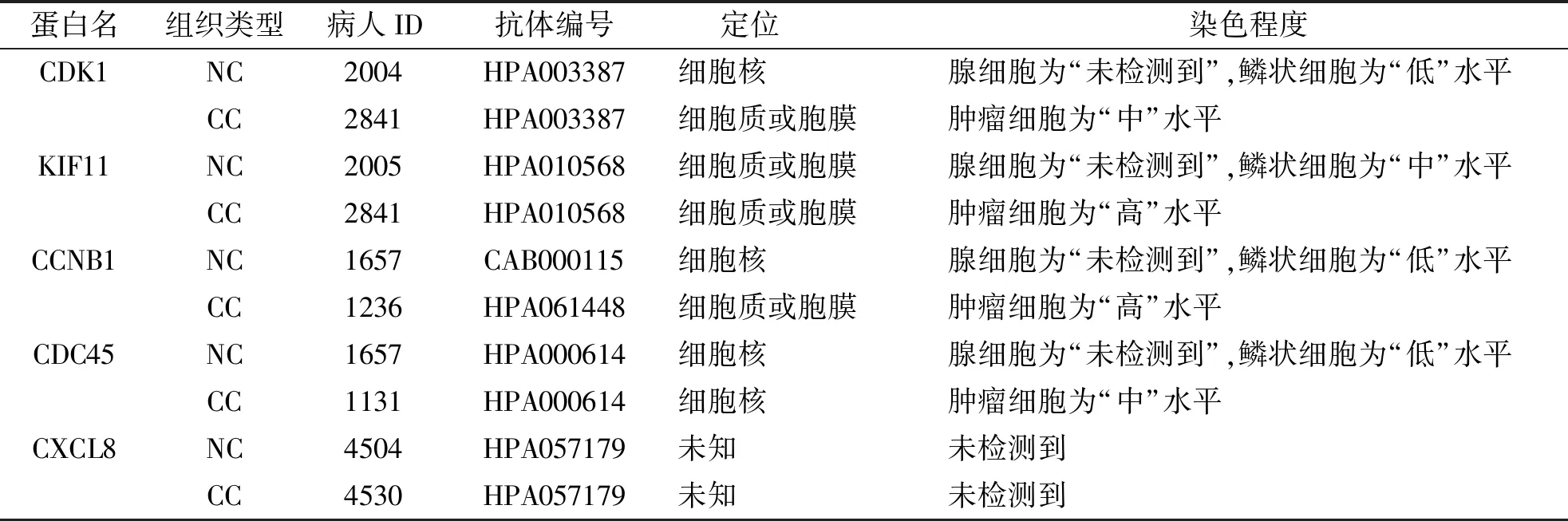
表5 CDK1、KIF11、CCNB1、 CDC45和CXCL8蛋白在NC及 CC肿瘤细胞中表达Tab.5 The expression of CDK1, KIF11,CCNB1,CDC45, and CXCL8 proteins in CC and NC tumor cells

图6 CC和NC组织相关蛋白的表达(免疫组织化学,×40)Fig.6 The expression of related proteins in CC and NC tissues(immunohistochemistry,×40)
3 讨论
CC是一种高度侵袭性肿瘤,是女性癌症相关死亡的主要原因之一,2018年全球估计有57万个新增病例,31.1万人死亡[1]。传统的治疗方式主要为手术和放疗,但中晚期CC单纯放疗效果差,患者5年生存率偏低,治疗效果仍不够理想[18]。因此,仍有必要为CC的诊断和治疗寻找新的靶点。CDK1是一种蛋白质编码基因,该基因编码的蛋白质是Ser/Thr蛋白激酶家族的成员[19]。该蛋白是高度保守的蛋白激酶复合物的催化亚基,被称为M期促进因子(maturation promoting factor,MPF),对于真核细胞周期的G1/S和G2/M相变至关重要[20]。CDK1已被确定为肺癌、乳腺癌和结直肠癌患者潜在的临床靶点和预后生物标志物[21]。CDK1在介导与CC进展相关的基因网络中起着全面的作用,靶向CDK1或其相关途径的新疗法可能有助于改善晚期CC的预后[22]。KIF11是驱动蛋白超家族的一员,这个蛋白质家族的成员已知参与各种纺锤体动力学,该基因产物的功能包括细胞有丝分裂过程中的染色体定位、中心体分离和双极纺锤体建立[23]。抑制KIF11能够引起细胞分裂紊乱和细胞周期阻滞,最终导致细胞凋亡,此外,KIF11能够调控轴突的分支和生长锥活性,研究表明KIF11在多种恶性肿瘤中高表达并与预后相关[24]。CCNB1基因编码的蛋白是一种参与有丝分裂的调节蛋白,其于正确控制细胞周期的G2/M转换期是必需的[25]。CCNB1与CDKl结合形成成熟促进因子MPF,MPF的激活是真核细胞启动有丝分裂必要条件,从而控制细胞周期进程[26]。既往研究发现CCNB1的高水平表达与肝癌、乳腺癌、胰腺癌及胃癌患者预后相关,其可能的机制多认为是抑制细胞增殖、迁移和侵袭,进而导致肿瘤的发生及发展[27]。CDC45编码的蛋白质是启动DNA复制所必需的蛋白质[28]。CDC45是高度保守的多蛋白复合体的成员,其在真核生物中DNA复制的早期步骤很重要[29]。染色质免疫共沉淀(chromatin immunoprecipitation, ChIP)实验发现CDC45与复制原点只在S期结合,同时这种结合需要CDK和CDC7的帮助,而CDC45在S期持续过程中远离复制原点[30]。CXCL8是CXC趋化因子家族的成员,是炎症反应的主要介质,负责中性粒细胞和粒细胞向炎症部位的招募和激活[31]。在对癌症的研究中,许多研究人员认为CXCL8在肿瘤的增殖、侵袭、迁移和肿瘤微环境中以自分泌或旁分泌的方式发挥着极其关键的作用[32]。在CC中,研究人员直接探讨CXCL8在组织和细胞系中的表达状况,并分析CXCL8表达与CC患者临床病理特征的关系[33]。但在本研究中,利用GEO数据库本研究筛选了CXCL8等基因作为CC的枢纽基因,并且在TCGA数据库中验证枢纽基因的表达去GEO数据库中一致,并探讨了枢纽基因对CC患者的总生存率的影响,这更有力地证明了CXCL8在CC中的预后功能。
本研究利用GEO数据库中CC表达微阵列GSE7803、GSE9750及GSE63514中的数据进行DEGs筛选,并对DEGs进行GO分析和KEGG通路分析,这些基因的GO富集主要包括有丝分裂细胞周期、细胞周期过程、丝氨酸型内肽酶活性等;KEGG信号通路主要富集在细胞周期、DNA复制、卵母细胞减数分裂、P53信号通路和花生四烯酸代谢。通过STRING及Cytoscape软件筛选出11个枢纽基因(CDK1、KIF11、BUB1B、CCNB1、CCND1、CDC20、CDC45、CXCL8、ECT2、ESR1及TOP2A),且利用The Kaplan Meier plotter网站分析得出5个枢纽基因(CDK1、KIF11、CCNB1、CDC45及CXCL8)CC患者总体生存率相关,进一步在TCGA数据库中验证了上述枢纽基因的表达水平,结果与GEO数据集的表达谱结果一致,并利用HPA数据库验证以上五种基因编码的蛋白在CC中较正常宫颈组织的表达情况,结果显示除CXCL8蛋白外,其余在肿瘤细胞中均呈表达上升水平。CXCL8 mRNA在CC中表达增高但CXCL8蛋白未被检测到的原因可能是因为转录后调控和翻译及翻译后调控,再有就是mRNA的降解、蛋白的降解、修饰折叠等因素导致mRNA丰度与蛋白表达水平不一致。最后综合分析发现CXCL8 mRNA 在CC患者中高表达且OS较差, 这表明高表达的CXCL8与CSCC的预后有关。
综上所述,本研究通过运用多种生物信息学分析方法筛选CC枢纽基因及信号通路,进一步对枢纽基因进行预后分析,挖掘CC预后分析的潜在分子标志,最终鉴定了5个CC枢纽基因,分别为CDK1、KIF11、CCNB1、CDC45及CXCL8,为CC治疗及预后分析提供新的思路。
