钴基氧还原电催化材料研究进展
2020-12-30张玉静蔡春蕾张慧娟
贾 立, 张玉静, 蔡春蕾, 耿 晶, 张慧娟
(上海理工大学 材料科学与工程学院,上海 200093)
阴极氧还原反应(oxygen reduction reaction,ORR)是燃料电池和金属-空气电池等可持续能源转换和储存系统的关键电极反应[1]。目前,使用最有效的是铂和铂基合金,但成本高和资源匮乏限制了其大规模应用[2]。为了降低ORR 催化剂的成本,促进燃料电池的商业化,研究者们一直在探索低成本非贵金属催化剂。本文综述了近年来钴基催化剂,包括Co 合金、Co-N-C,Co 纳米离子、钴基硫化物、钴基磷化物和钴基其他形式,用于ORR 催化剂的研究进展,并在此基础上对未来高效、廉价、稳定、环保的新型氧还原催化剂的设计进行了展望。
1 Co 合金
与单一金属催化剂相比,合金纳米粒子由于其独特的电子效应和协同效应,表现出更高的活性。Dai 等[3]设计了一种三元Co@Pd-Pt 催化剂,即Pt三聚体(Pt3)修饰在Co-Pd(Co@Pd)核壳上,Pt 负载量为2.4%,在碱性电解液中其Co@Pd-Pt/CNT 的氧还原起始电位为0.944 V,比商业Pt/C(0.956 V)的稍低,但其质量活性比Pt/C 的提高了30.6 倍,且具有良好的稳定性,322 000 圈循环后仍保持较高的活性。Han 等[4]通过热分解含有多巴胺(dopamine,DPA)涂层的金属有机框架(metal organic frameworks,MOFs)合成一种氮掺杂空心碳纳米立方体负载原子级二元Co-Ni 位(CoNi-SAs/NC),如图1 所示。其起始电位(0.88 V)、半波电位(0.76 V)和极限电流密度( 4.95 mA/cm2) 与 商 用Pt/C( 0.90 V, 0.82 V,5.09 mA/cm2)的相近。在16 h 计时电流后,CoNi-SAs/NC 的稳定性(90%)远高于Pt/C(80%)的,且具有良好的抗甲醇性能。Ying 等[5]利用一种Co 基金属有机骨架制备PtCo 双金属纳米颗粒,并与含氮空心多孔碳包覆的Co 纳米混合得到PtCo/Co@NHPCC, 其 质 量 活 性 和 比 活 性( 0.566 A/mgPt,0.876 mA/cm2)优于商用Pt/C 催化剂(0.102 A/mgPt,0.177 mA/cm2) 的 和 商 用 Pt 黑( 0.042 A/mgPt,0.221 mA/cm2)的,且稳定性好,在5 000 次电位扫描后,催化活性几乎没有变化。Tan 等[6]以Pt-25at%Co和纯Pt 为基准,系统地研究了Pt-Au-Co 和Pt-Ir-Co 薄膜合金的ORR 催化活性和稳定性。Pt-2.5%Au-25%Co 合金在0.95 V 下的比活度为1.41 mA/cm2,分别比Pt-Co 和纯Pt 的高16%和404%,增加Au 或Ir,合金的稳定性显著提高,Pt-10%Au-25%Co 在100 000 圈循环后,比活度仅损失25%,而Pt-25%Co 的损失80%。

图 1 CoNi-SAs/NC[4]Fig.1 CoNi-SAs/NC[4]
2 Co-N-C
Co-N-C 被认为是最有前途的ORR 催化材料。直接热解碳载体,含氮前驱体和钴盐的混合物是目前被广泛采用的一种制备方法,但不能很好地控制活性中心,而且常常导致严重的聚集,这对ORR 的性能有3 个方面的负面影响:(1)不可逆聚集大大减少了表面活性位点,从而限制了活性;(2)粗热解制备的催化剂比表面积低,孔结构差,阻碍了反应物进入活性中心;(3)活性中心分布的弱操纵降低了催化剂的重现性。开发一条比表面积高、孔结构丰富、活性中心分布均匀的Co-N-C 催化剂的制备方法迫在眉睫。
Chen 等[7]以三聚氯氰和哌嗪为单体溶液缩聚法合成富氮共价有机聚合物(covalent organic polymers,COP),以钴离子配位的COP(Co-COP)为前驱体,采用简单热解法合成Co/N-PCNF,具有良好的导电性、层次多孔结构和均匀分布的活性中心,且具有优异的ORR 催化活性、稳定性和抗甲醇性能,结果表明其具有近四电子的氧还原途径,半波电位为0.835 V,与Pt/C 催化剂(0.865 V)的相当;连续运行40 000 s 后,电流密度保持在94%;加入1 M甲醇后,催化性能没有明显变化。Han 等[8]制备了由Co 和N 双掺杂碳纳米片和碳纳米管组成的3DCo@N/C 催化剂,其起始电位为0.915 V,与Pt/C(0.97 V)的相当,且具有较高的半波电位(0.812 V),仅略低于Pt/C 的半波电位(0.856 V)。这种三维结构具有高比表面积且能充分暴露活性位点,吡啶类N、Co-NX和Co 纳米粒子的强界面接触具有协同效应,从而显著提高了其电催化性能。Park 等[9]通过设计核壳型杂化MOFs(ZIF-L@ZIF-67)一步热解制备了Co、N 共掺多孔炭叶(Co, N-PCLs),具有较正的半波电位、较大的扩散电流密度、四电子转移、良好的稳定性和甲醇耐受性,这些优异的性能归因于其独特的结构和组成特征,如扁平的形貌、高比表面积/孔隙率、大量石墨碳(包括碳纳米管)以及均匀的Co 和N 掺杂。Hu 等[10]以聚苯乙烯@聚多巴胺为原料,采用热解法制备了一种新型的Co-N 掺杂介孔碳空心球(Co-N-mC),Co 活性位点广泛分布在高度石墨化的介孔N 掺杂碳空心球中,在0.1 M KOH 溶液中表现出优异的ORR 活性,起始电位为0.940 V,半波电位为0.851 V,Tafel 斜率为45 mV/dec,与20%Pt/C 催化剂的相当,且在60 000 s 稳定性测试中,电流几乎没有下降(<4%),这些特性使Co-NmC 成为迄今为止在碱性条件下用于ORR 的最佳非贵金属催化剂之一。
3 Co 纳米粒子
金属纳米粒子的团聚和尺寸过大可能严重影响催化剂的氧还原反应活性,与氧化物和氧氢氧化物等相比,零价过渡金属在ORR 催化中常常被忽略。研究表明,即使碳中没有氮掺杂,零价钴也能有效地提高邻近碳的ORR 活性。
Hou 等[11]在水溶液中成功地制备了正丁胺脱质子诱导的ZIF-67 纳米笼及其衍生的钴纳米粒子包埋Co,N-共掺碳空心纳米笼(Co@NPC-H),结果表明,Co@NPC-H 的起始电位为0.88 V,氧还原峰位为0.76 V,极限电流密度为-5.20 mA/cm2,与商业Pt/C 的 相 当( -5.00 mA/cm2) , 塔 菲 尔 斜 率(33.2 mV/dec)比商用Pt/C 的(46.0 mV/dec)低,且具有更优异的稳定性和耐甲醇性能。Guo 等[12]制备了空心氮/钴掺杂多孔碳胶囊负载钴纳米颗粒(Co@CoNPCC),如图2 所示,在0.1 M KOH 中,其起始电位为1.02 V,电流密度为5.2 mA/cm2。Liu等[13]在Co@NPC 中研究了酸洗时间对Co 纳米粒子的影响,结果表明,Co@NPC-APt(t=12 h)具有比Co@NPC 更好的电催化ORR 活性,Co@NPC-APt(t=12 h)的极限扩散极限电流密度为-5.6 mA/cm2远 高 于 Co@NPC ( -4.7 mA/cm2) 的, 与 Pt-C(-5.6 mA/cm2)的相近;在0.6 V 下Co/NPC-AP 的动力学电流密度值为166 mA/cm2,约为Co@NPC(50 mA/cm2)的2.6 倍,与Pt-C 的催化性能相近。Liu 等[14]采用水热法合成了金属钴纳米粒子修饰石墨烯气凝胶,并在H2气氛下进行了热退火,制备钴纳米粒子修饰还原石墨烯(CoNPs/rGO),结果表明,其半波电位和四电子选择性都非常接近Pt/C 的,且具有优异的电化学耐久性。
4 Co 基氧化物
过渡金属及其化合物在ORR 中表现出优异的电催化活性,其中Co 基氧化物是研究最为广泛的材料之一。精细纳米结构是创造更大表面积和暴露更多表面原子的有效途径,且往往伴随着更多的缺陷和更丰富的微孔,从而促进电荷迁移,提高催化剂活性。因此,人们一直致力于合成各种结构的CoxO 材料,包括Co3O4纳米颗粒、纳米线、纳米薄膜和花状结构等。
Chen 等[15]通过直接氧化碳包覆的Co 纳米粒子,制备了中空Co3O4纳米笼修饰三维石墨烯气凝胶(Co3O4/3DG),具有较高的起始电位(0.82 V)、较大的极限电流密度(5.12 mA/cm2)、电子转移数约为4、优良的耐甲醇性能和耐用性。He 等[16]采用简单的一步溶剂热法合成了多壁碳纳米管负载的钯钴氧化物纳米粒子(Pd-CoO/MWCNTs),CoO 的引入提高了Pd-CoO/MWCNTs 的比活性,其电流密度为5.29 mA/cm2,接近于20%Pt/C(5.37 mA/cm2)的催化性能。稳定性结果表明,Pd-CoO/MWCNTs 的半波电位几乎没有变,起始电位只负移12 mV,相比之下,20%Pt/C 的起始电位降低了55 mV,且其半波电位降低了41 mV,说明引入CoO 纳米粒子可以进一步提高催化剂的稳定性和对氧还原反应的催化活性。Liu 等[17]将CoO 纳米颗粒嵌入氮硫共掺碳纳米纤维制备了一种新型的三维双功能电催化剂(CoO@N/S-CNF)。研究表明:CoO@N/S-CNF的起始电位约为0.84 V,且在0.4 V 下电流密度为5.1 mA/cm2,接近20%Pt/C 的电流密度,稳定性测试表明CoO@N/S-CNF 电流损失为20%,而Pt/C 的电流损失接近50%,表明CoO@N/S-CNF 具有更好的稳定性,这归因于CoO 纳米粒子和碳纳米纤维以及氮硫共掺杂的双重协同效应。Wang 等[18]通过改变Co3O4/聚苯胺的热解温度调节Co 在CoOx@NG 中的氧化状态,聚苯胺在高温下分解释放气体还原Co3O4,同时作为原位生长石墨烯的前驱体,纯聚苯胺制备的NC-800 的性能远不如CoOx@NG-800 的,说明CoOx 核在调节碳电子结构中的重要作用。
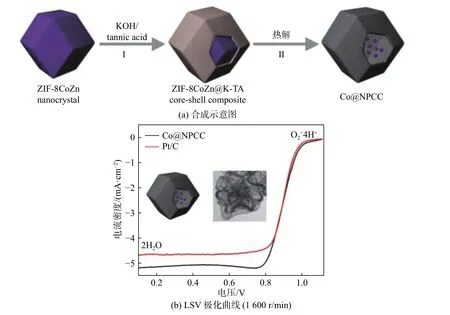
图 2 Co@CoNPCC[12]Fig.2 Co@CoNPCC[12]
5 Co 基磷化物
近年来,包括CoP、CoP2和Co2P 在内的亚磷酸钴(CoxP)作为新兴的析氢反应(hydrogen evolution reaction,HER)电催化剂引起了研究人员的关注。有少量的研究表明,亚磷酸钴也表现出优异的OER 活性[14]。然而,对ORR 的电催化活性却鲜有研究。最近,基于类海胆CoP 纳米晶的HER 和ORR 双功能催化剂才被制备出来,但对ORR 的电化学活性仍有待提高。事实上,半导体CoxP 的低导电性严重阻碍了其电化学性能。为了克服这一障碍,高比表面积、高导电性和超稳定性的碳载体如多孔炭、碳纳米管和石墨烯是一种可行的措施。
Wang 等[19]研究了一种由纳米Co2P、N、P 共掺杂碳(NPC)和CeO2纳米片组成的三元催化体系Co2P-NPC-CeO2,其中CeO2纳米片被多巴胺衍生的碳层包裹,植酸作为磷源用Co2P 纳米粒子修饰,催化剂的起始电位为0.88 V,半波电位为0.827 V,扩散电流密度为5.24 mA/cm2,1 000 次循环后半波电位无变化,其主要是由于CeO2具有良好的氧吸附性能,Co2P 中暴露的活性中心较多,氮磷共掺杂碳层具有良好的导电性,且Co2P 中的电子会转移到CeO2中,导致大量的氧空位。Lv 等[20]通过原位水热处理制备了还原氧化石墨烯负载CoP(rGO@CoP),其起始电位为-0.148 V(vs.SCE);与纯CoP 相比,增加了0.129 V,且在电流密度为1.0 mA/cm2时电位也增加了0.330 V;在电位为-0.350 V(vs.SCE)时增加了0.51 mA/cm2,塔菲尔斜率下降了19 mV/dec,电子转移数为3.66,大于纯CoP 的2.19。如图3 所示,Jiang 等[21]通过一种简单的超分子凝胶辅助热解方法,合成了Co2P 锚定在钴、氮、磷共掺杂石墨烯纳米片Co2P@CoNPG-900,起始电位(0.90 V)和半波电位E1/2(0.81 V)均比Pt/C 的高10 mV,且极限电流密度(6.68 mA/cm2)比Pt/C 催化剂(5.67 mA/cm2)高得多,其优异的催化性能可归因于Co2P 和Co-Nx 活性中心的协同耦合效应以及氮磷共功能化石墨烯提供众多的缺陷点。
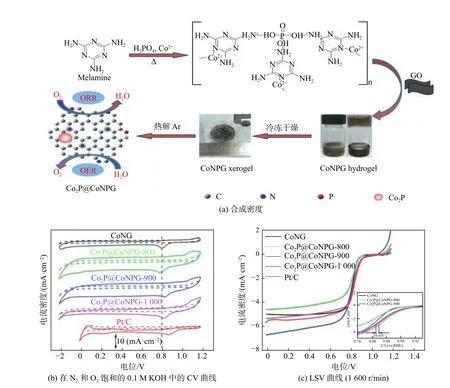
图 3 Co2P@CoNPG[21]Fig.3 Co2P@CoNPG[21]
6 Co 基硫化物
钴基硫化物因其具有优异的电化学活性而受到广泛关注,例如,Co9S8,Co3S4和Co1-xS 对ORR 和OER 都具有优异的性能;此外,CoS2也是一种很有前途的ORR 电催化剂,然而,CoS2的催化活性因其导电率低而受到限制。一般来说,在石墨烯表面生长或支撑硫化钴纳米颗粒是提高其导电性的有效途径。
Feng 等[22]以ZIF-67 MOFs 为前驱体,制备了氮掺杂多孔石墨化碳载二硫化钴(CoS2@NC),如图6 所示,其起始电位为-0.105 V(vs.Ag/AgCl),半波电位为-0.172 V(vs.Ag/AgCl),扩散极限电流密度为5.49 mA/cm2,与商用Pt/C 的性能相当,该方法是一种简单有效的合成氧自由基碳担载金属硫化物的方法,多孔结构与纳米晶粒的结合能有效地缩短离子的输运路径,增加活性区,从而大大提高电极材料的导电性,提高其ORR 催化活性。Qiao 等[23]通过简单的一锅热解法将硫化钴空心纳米球嵌入氮和硫共掺杂的石墨烯纳米孔中Co1-xS/N-S-G,具有分级介孔结构,在碱性介质中,该催化剂表现出半波电位比商用Pt/C 的高30 mV,扩散极限电流密度比商用Pt/C 的高15%。研究表明,该材料优异的电催化性能归因于:(1)多种活性中心,如硫化钴空心纳米球、氮、硫掺杂原子和Co-N-C 中心,引起的协同效应;(2)硫化钴空心纳米球穿过石墨烯的平面,形成了它们之间的强相互作用;(3)石墨烯上纳米孔的存在导致了更多的边缘缺陷;(4)多层多孔结构产生了高的比表面积和良好的传质速率。
Cao 等[24]通过将吡咯与Co9S8纳米粒子聚合再热解的方法制备了一种Co9S8/N-C,其起始电位是-0.05 V(vs.Ag/AgCl);与商用Pt/C 的相比,半波电位仅相差39 mV,在低过电位下,Tafel 斜率为69 mV/dec 与商用Pt/C(65 mV/dec)的接近。此外,在-0.2 V(vs.Ag/AgCl)电位下连续运行90 万次后电流密度仍为95%,相比之下,商业Pt/C 的电流密度下降了59%。结果表明,Co9S8与N-C 之间的共价偶联作用和高比表面积是形成该材料具有高活性和高耐久性的重要原因。
7 Co 基其他形式
Lai 等[25]通过原位热解Co/Zn-ZIF-67 成功地制备了CoN3纳米颗粒。在热解过程中,Co/Zn-ZIF-67首先转化为N 掺杂多孔碳负载Co 纳米粒子Co@NC,并伴随着ZIF 结构分解释放NH3,Zn 蒸发形成的大量微孔和Co@NC 的大表面积促进了NH3分子与Co 接 触,生 成了CoN3,CoN3@NC-7-1 000 显 示 出优异的活性和稳定性,在0.5 M H2SO4溶液中具有较小的正半波电位(0.72 V)和高电流密度(5.40 mA/cm2)。密度泛函理论计算表明,CoN3的(220)面对O2分子具有较低的吸附势垒,能有效地促进酸性电解质中的ORR 性能。此外,以CoN3@NC-7-1 000 为阴极催化剂组装的H2/O2燃料电池具有良好的催化活性,峰值功率密度为160 mW/cm2,且稳定性好。Zhen 等[26]制备了Mn 掺杂氮氧化钴与氮掺杂还原氧化石墨烯纳米片杂化物CoMnON/N-rGO。通过掺入Mn 并与N-rGO 偶联,可以显著提高电荷转移率和比表面积。测试表明,Mn 掺杂后CoMnON/N-rGO 的起始电位为0.91 V,半波电位为0.83 V,且在1.0 M KOH 溶液中的Tafel 斜率为70 mV/dec,Mn 的加入、二维介孔结构与N-rGO 纳米片的耦合作用,均提高了材料的比表面积和活性表面位。
Zhou 等[27]在碱性介质中合成了一种磷酸化金属有机骨架磷酸钴杂化碳前驱体,如图4 所示。通过热解制备了氮配位磷酸钴碳杂化材料Co3(PO4)2C-N/rGOA,结果表明,其在0.1 M KOH 电解液中的起始电位为0.962 V,半波电位为0.837 V,与Pt/C 的性能相当,且在碱性介质中具有良好的稳定性和抗甲醇性能,这归因于氮掺杂石墨碳、含碳磷酸钴和具有适当比表面积的石墨烯的协同作用。
8 展 望
本文总结了Co 基ORR 催化剂的研究进展,讨论了不同形式Co 基材料的合成方法以及其催化性能,主要分为Co 合金、Co-N-C、Co 纳米粒子、Co 基氧化物、Co 基磷化物和Co 基硫化物6 大类。Co 基材料能够提高催化活性的原因有:(1)多种活性中心,暴露出更多的活性位点;(2)构造三维多孔的结构,形成高的比表面积和良好的传质速率;(3) 改变催化剂的电子结构,优化其本征活性;(4)提高电极材料的导电性,从而实现电子的快速运输。目前,已有Co 基材料催化剂的ORR 催化活性能堪比Pt 等贵金属催化剂的,有望成为新一代高效、廉价、稳定的氧还原反应催化剂。但是,上述进展多数还处于实验室研究阶段,进一步开发成本低、催化活性高、稳定性好的催化剂还需要大量的研究,如发展高效低成本的合成方法,从理论上探究催化剂的“构-效”关系等,从而更好的指导高性能低成本的ORR 催化剂的设计与合成。
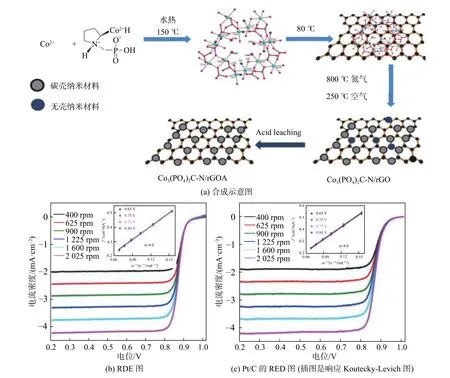
图 4 Co3(PO4)2C-N/rGOA[27]Fig.4 Co3(PO4)2C-N/rGOA[27]
