复方蒲公英颗粒剂的质量标准研究
2020-12-25张玉爱郭智强廖太祥陈圆黎莉萍南昌市博泽康医药科技有限公司南昌330029
★ 张玉爱 郭智强 廖太祥 陈圆 黎莉萍(南昌市博泽康医药科技有限公司 南昌 330029)
复方蒲公英颗粒是由蒲公英、黄连、薏苡仁、茵陈等十二味中药材制成的复方制剂,具有清泄肺胃积热、解毒散结等功效。适用于湿热毒蕴结所致的痤疮,脂溢性皮炎,玫瑰痤疮,酒渣鼻,毛囊炎等病症。原中药处方没有固定的质量标准,很难对药品质量进行控制,为了保障在临床中应用的安全,因此有必要进行相关的医院制剂质量标准研究[1-2]。
本文对该处方药物进行质量标准研究,试验采用TLC法对处方中的蒲公英、黄连和薏苡仁进行了定性鉴别,并以盐酸小檗碱为定量指标,研究了HPLC测定该成分的方法,并进行方法学考察,最终建立起复方蒲公英颗粒的质量标准,保证临床用药的安全、有效和稳定。
1 仪器与材料
1.1 仪器 KQ2200DB型超声波清洗机(昆山超声波仪器有限公司);ZF-2型紫外仪(上海安亭电子仪器厂);BS25S型电子天平(奥豪斯仪器有限公司);APS-80-10D型高效液相色谱仪(德国沃特斯);硅胶G板(青岛海洋化工厂)。
1.2 材料 盐酸小檗碱对照品(批号:110713-200208,中国药品生物制品检定研究院);蒲公英对照药材(批号:121195-201503,中国药品生物制品检定研究院);薏苡仁对照药材(批号:121254-201504,中国药品生物制品检定研究院);乙腈为色谱纯;水为超纯水(自制);其它化学试剂均为分析纯。复方蒲公英颗粒供试品三批(170504、170505、170506)及阴性样品均由本公司制剂室自制。
2 方法与结果
2.1 薄层色谱法
2.1.1 黄连的鉴别 黄连中的主要化学成分为盐酸小檗碱[3],故以盐酸小檗碱进行定性鉴别。①供试品溶液:取本品10 g,研细,加乙醇30 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。②对照品溶液:再取盐酸小檗碱对照品,加甲醇制成每1 mL含0.5 mg的对照品溶液。③黄连阴性对照品溶液:再取缺黄连药材的复方蒲公英颗粒内容物10 g,按供试品溶液制备的方法制成阴性对照品溶液。④检查方法:照薄层色谱法(中国药典2015年版四部通则0502)试验[4],吸取供试品溶液5 μL、阴性对照品溶液5 μL、对照品溶液2 μL,分别点于同一硅胶G薄层板上,以正丁醇-冰醋酸-水(7∶1∶2)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。⑤结果:供试品溶液色谱中,在与对照品溶液色谱相应的位置上显相同的黄色荧光斑点;阴性对照色谱中,无上述斑点检出。说明本法专属性强,可以作为黄连的鉴别。

图1 黄连薄层鉴别图谱
2.1.2 蒲公英的鉴别[5]①供试品溶液:取本品10 g,研细,加乙醇30 mL,超声处理20 min,滤过,滤液蒸干,残渣加三氯甲烷2 mL使溶解,作为供试品溶液。②对照药材溶液:另取蒲公英对照药材1 g,加甲醇20 mL,同法制得对照药材溶液。③蒲公英阴性对照品溶液:再取缺蒲公英药材的复方蒲公英颗粒内容物10 g,按供试品溶液制备的方法制成阴性对照品溶液。④检查方法:照薄层色谱法(中国药典2015年版四部通则0502)试验[4],吸取供试品溶液、阴性对照品溶液与对照药材溶液各10~15 μL,分别点于同一硅胶G薄层板上,以三氯甲烷为展开剂,展开,取出,晾干,喷以10 %硫酸乙醇溶液,加热至斑点显色清晰。⑤结果:供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点;阴性对照色谱中,无上述斑点检出。说明本法专属性强,可以作为蒲公英的鉴别。如图2所示。

图2 蒲公英薄层鉴别图谱
2.1.3 薏苡仁的鉴别[6]①供试品溶液:取本品10 g,研细,加石油醚(60~90 ℃)20 mL,超声处理30 min,滤过,滤液蒸干,残渣加石油醚1 mL使溶解,作为供试品溶液。②对照药材溶液:另取薏苡仁对照药材1 g,同法制得对照药材溶液。③薏苡仁阴性对照品溶液:再取缺薏苡仁药材的复方蒲公英颗粒内容物10 g,按供试品溶液制备的方法制成阴性对照品溶液。④检查方法:照薄层色谱法(通则0502)试验[4],吸取供试品溶液、阴性对照品溶液与对照药材溶液各5~10 μL,分别点于同一硅胶G薄层板上,以石油醚-乙酸乙酯-醋酸(10∶3∶0.1)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。⑤结果:供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点;阴性对照色谱中,无上述斑点检出。说明本法专属性强,可以作为薏苡仁的鉴别。
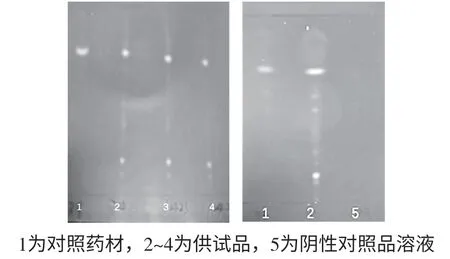
图3 薏苡仁薄层鉴别图谱
2.2 复方蒲公英颗粒检查
2.2.1 水分 按照《中国药典》2015年版四部通则0832水分测定法测定,取同一批复方蒲公英颗粒,平行3份,测得平均含水量为4.32 %(小于8.0 %),符合要求。
2.2.2 溶化性 按照《中国药典》2015年版四部通则0104颗粒剂溶化性项下规定,取复方蒲公英颗粒10 g,加热水200 mL,搅拌5 min,观察到颗粒全部溶化,符合要求。
2.2.3 粒度 按照《中国药典》2015年版四部通则0982粒度和粒度分布测定法第二法双筛分法测定,结果不能通过一号筛与能通过五号筛的颗粒总和为7.0 g(不超过15 %)。
2.2.4 装量差异 按照《中国药典》2015年版四部通则0104颗粒剂装量差异项下规定(6.0 g以上,装量差异限度为±5 %),复方蒲连颗粒三批样品装量差异检测结果均在±5 %范围内,符合药典项下规定。
2.2.5 微生物的检查方法验证 根据实际检验需求,从需氧菌总数计数法、霉菌和酵母菌总数计数法和控制菌检查方法三个方面对复方蒲连颗粒微生物检查方法进行了详细具体的验证,验证结果均符合要求,证明了复方蒲连颗粒微生物检查方法是合理有效的,所获得的检验结果也是真实可靠的。
2.3 盐酸小檗碱的含量测定
2.3.1 色谱条件[4]色谱条件:Diamonsil C18色谱柱(250mm×4.6mm,5μm);流动相为乙腈-0.05 mol/L磷酸二氢钾溶液(50∶50)(每100 mL中加十二烷基硫酸钠0.4 g,再以磷酸调节pH值为4.0)为流动相;检测波长为345 nm;进样量为20 μL。在此色谱条件下,供试品中的盐酸小檗碱与其他成分有较好的分离度,阴性样品对指标成分含量测定无干扰。

图4 高效液相色谱图
2.3.2 对照品溶液的制备 取盐酸小檗碱对照品适量,精密称定,加甲醇制成每1 mL约含20 μg的溶液,即得。
2.3.3 供试品溶液的制备 取装量差异项下的本品,研细,取约5 g,精密称定,置具塞锥形瓶中,精密加入甲醇:盐酸(100∶1)混合溶液20 mL,密塞,称定重量,超声处理30 min,放冷,再称定重量,用上述混合溶液补足减失的重量,摇匀,滤过(0.45 μm滤膜),取续滤液,即得。
2.3.4 阴性对照溶液的制备 按处方比例,称取缺少黄连的复方蒲公英颗粒的药材,按照制备工艺配制,然后依照“2.3.3”项下的方法制备阴性对照溶液。
2.3.5 线性关系考察 精密称取盐酸小檗碱对照品33.0 mg,置50 mL量瓶中,加甲醇稀释至刻度,摇匀,制成0.66 mg/mL的对照品贮备液。精密量取对照品贮备液0.5、1、2、3、4、5、10 mL,置10 mL量瓶中,加甲醇稀释至刻度,摇匀。吸取上述溶液,分别进样20 μL,以测定浓度X(mg/mL)对峰面积Y进行线性回归,得回归方程Y=4×107X-50 694,r=0.999 6(n=7),试验结果表明盐酸小檗碱在0.003 42~0.068 40 mg/mL范围内呈良好线性关系。
2.3.6 精密度试验 取盐酸小檗碱对照品溶液(20 μg/mL)20 μL,连续进样6次,测定峰面积,结果对照品峰面积RSD=1.66 %,精密度良好。
2.3.7 重复性试验 取本品同一批号样品6份,按2.3.3项下方法操作,分别按上述色谱条件方法进行测定盐酸小檗碱,计算样品含量,RSD为2.20 %(n=6),表明该含量测定方法有良好的重复性。
2.3.8 稳定性试验 取同一批号样品溶液(170504),室温放置,在0、2、4、6、8、12、24 h分别精密吸取20 μL测定,按上述色谱条件测定结果峰面积的RSD为1.26 %。色谱峰面积在24 h内基本无变化,说明稳定性好。
2.3.9 回收率试验 取已知含量的同一批号(170504)样品6份,研碎,分别加入盐酸小檗碱对照品溶液(0.142 0 mg/mL)1 mL,按上述2.3.3项下制备方法配制加样回收供试液,按上述色谱条件测定,计算回收率,结果见表1。

表1 盐酸小檗碱回收率试验结果
2.3.10 样品含量测定 分别精密吸取对照品溶液与供试品溶液各20 μL,注入液相色谱仪,测定峰面积,按外标法计算3批中试样品(170504、170505、170506)含量。

表2 三批样品中盐酸小檗碱含量测定结果
3 讨论
3.1 专属性 本试验对处方中的黄连、桑白皮、大黄、蒲公英、薏苡仁、茵陈等进行了薄层鉴别条件摸索,发现桑白皮、大黄、茵陈等均有阴性干扰,因此确定了黄连、蒲公英和薏苡仁作为薄层鉴别方法,重现性好,结果可靠。同时参考中国药典2015年版一部收载的黄连中盐酸小檗碱含量测定方法,通过方法学验证,确定了本品的含量测定方法,方法简便,稳定,快捷,且所测3批样品中盐酸小檗碱的含量稳定,方法可行。
3.2 提取方法的选择 对加水量、提取时间、提取次数进行了摸索及确定,进行了三因素三水平正交实验,结果表明,提取次数对提取工艺影响最大,其次是加水量,提取时间对提取工艺影响较小。从经济成本、实际生产考虑,确定本品提取最佳工艺条件为:加水量为8倍,提取时间为2 h,提取次数为2次。
3.3 浓缩工艺考察 日常生产过程中,我们考察滤液浓缩的相对密度对制剂成型工艺的影响,通过将制粒过程及颗粒的性状作为参考指标,根据实验结果,发现滤液浓缩的相对密度为1.35最适宜,考虑到实际生产的可操作性,将滤液浓缩的相对密度定为1.32~1.35(60 ℃)的稠膏。
3.4 制剂工艺的选择 日常生产过程中,中药颗粒剂一般采用蔗糖作为矫味剂,糊精、淀粉作为粘合剂,我们分别对其进行了考察,并以溶化性作为参考依据,结果发现蔗糖加糊精,溶化完全,无分层现象;蔗糖加淀粉,溶化性不好,出现少量分层现象。故根据实验结果,采用蔗糖、糊精作为制剂成型辅料。根据实验结果,加入蔗糖与糊精比例为2∶3,软材适中,颗粒均匀。由于实际生产过程中药材批次不同,出膏率不一样,可进行适当微调。
3.5 干燥工艺的选择 日常生产过程中,我们考察干燥工艺对制剂成型工艺的影响,通过将不同干燥温度与干燥时间来测定水分作为参考指标,实验结果显示,均符合2015年版《中国药典》规定(8.0 %<水分),根据实际生产情况,将干燥温度定为60~80 ℃,干燥时间定为2~4 h,考虑到生产批量不同,可适当微调。
3.6 检测波长的选择 采用二极管阵列检测器对盐酸小檗碱对照品色谱峰进行光谱扫描,结果盐酸小檗碱在345 nm波长处有最大吸收,由于345 nm杂峰对目标峰干扰少且稳定,故选择345 nm作为检测波长。
3.7 色谱条件的优化 本实验通过摸索乙腈-0.05 mol/L磷酸水溶液、甲醇-0.05 mol/L磷酸水溶液按一定比例混合作为流动相,考察其分离度,结果以乙腈-0.05 mol/L磷酸二氢钾溶液(50∶50)(每100 mL中加十二烷基硫酸钠0.4 g,再以磷酸调节pH值为4.0)为流动相;检测波长为345 nm,Diamonsil C18色谱柱(250 mm×4.6 mm,5 μm)的色谱条件下供试品中盐酸小檗碱与其他共存组分峰能达到良好的分离。
本实验建立了复方蒲公英颗粒中盐酸小檗碱的HPLC含量测定方法。结果表明,所建立的方法操作简便、结果可靠、重复性好。
