不同体系痕量磷检测及其在磷价态转化中的应用
2020-12-16毕明洋黄国强
毕明洋,黄国强
(天津大学化工学院,天津 300350)
三氯氢硅是制备半导体用高纯度硅的基本原料,对于这种原料的杂质控制至为重要[1],杂质主要以硼磷金属为主,痕量磷对于硅的电学性能有显著的影响,其在三氯氢硅中主要以PCl3、PCl5和POCl3形式存在,尽管三氯氢硅中痕量磷去除手段研究广泛,但是很少有系统的方式对测定三氯氢硅中的痕量磷的方法进行研究,因为三氯氢硅性质特殊,腐蚀性强极易挥发,会对仪器灵敏度及使用寿命产生严重影响[2-3],所以三氯氢硅精制除磷的研究可以采用与三氯氢硅性质相似的有机溶液代替。
迄今为止,国内外对痕量磷杂质的检测主要有紫外-可见光分光光度计法、原子吸收光谱法、极谱法、电感耦合等离子体发射光谱法、电感耦合等离子体质谱法和离子色谱等,其中电感耦合等离子体质谱法和电感耦合等离子体发射光谱法占据主流,但大多数研究都没能解决其对操作环境、样品性质要求较高,稳定性差,无法普及的缺点[4];关于原子吸收光谱、极谱法和离子色谱法系统研究的公开数据很少。
本研究讨论了紫外-可见分光光度计法测定不同体系痕量磷的方法,此处利用高纯水萃取分离操作来提取有机溶剂中痕量磷杂质,它比起其他方法具有操作较简单,技术成熟,价格低,分析速度快,选择性高等一系列优点。通过氧化差减法改进后的分光光度计法可以同时检测水溶液中的磷酸和亚磷酸,并配合萃取分离实现不同体系痕量磷的测定。而后对有机溶剂中磷杂质价态转化效果进行了测定,聚合物负载铬氧化剂能在一定条件下将杂质中的PCl3氧化为POCl3,更强的给电子能力,将大大提升络合吸附和精馏的脱除效果[5]。
1 实验部分
1.1 材料与仪器
UV1800PC型紫外分光光度计,上海丙林电子科技有限公司;高纯水发生机,东莞市谦和水处理有限公司;X射线衍射仪(Bruker铜靶),德国布鲁克AXS有限公司;实验所用试剂均为分析纯。
1.2 特殊溶液的配制
混合发色剂:按序量取5 mol/L硫酸溶液、质量分数为3%的钼酸铵溶液、质量分数为5.4%的抗坏血酸溶液和质量分数为0.136%的酒石酸氧锑钾溶液于1个容器中,体积比按照3∶2∶2∶1配比,每加1种后均需混匀,盛于试剂瓶中。此混合溶剂有效时间6 h,临使用前适量配置。
5价磷标准使用溶液:称取1.088 g KH2PO4,溶于高纯水,转移至1 000 mL容量瓶,稀释至标线,混匀;移取1.00 mL于100 mL容量瓶中,稀释至标线,混匀。此溶液1 mL含0.080 0 μmol 5价磷原子,有效期1周。
3价磷标准使用溶液:称取0.656 g H3PO3,溶于高纯水,转移至1 000 mL容量瓶稀释至标线,混匀; 移取1.00 mL于100 mL容量瓶中,稀释至标线,混匀。此溶液1 mL含0.08 μmol 3价磷原子,有效期1周。
1.3 对钼酸铵分光光度法的线性检测
钼酸铵分光光度法:在50 mL比色管中分别加入待测溶液,稀释至50 mL,混匀;各加入5 mL混合发色剂,混匀。15 min后,用1 cm比色皿在分光光度计上,从波长600~1 100 nm进行波长扫描,测其紫外吸收光谱,得出最大吸收波长出的吸光度A;通过由标准溶液绘制出的标准曲线计算出比色管中磷酸的浓度,从而计算出待测溶液中磷酸的量[6]。
对不同量的5价磷标准使用溶液以钼酸铵分光光度法测定其磷酸的量,并绘制标准曲线。
1.4 对氧化差减法测定亚磷酸浓度的线性检测
氧化差减法:在2个50 mL比色管A和B中,加入相同量的同一待测溶液,比色管A中加40 mL水,加入适量过硫酸钾饱和溶液,120 ℃加热一段时间,冷却后稀释至50 mL,混匀;比色管B中加入高纯水稀释至50 mL,混匀;在比色管A和B中均加入5 mL混合发色剂,发色15 min,用1 cm比色皿,在891 nm波长上测定吸光度A1和A2。A1和A2的差值在钼酸铵分光光度法标准曲线对应的磷酸浓度即为待测溶液中亚磷酸的浓度[7]。
对不同量的3价磷标准使用溶液以氧化差减法测定其亚磷酸的量,并绘制工作曲线。
1.5 不同条件下5价磷元素在正己烷-水萃取体系中萃取率的影响因素
将不同量的POCl3溶解于不同量的正己烷中,溶解完全后在分液漏斗中用50 mL高纯水室温下萃取20 min,放出下层水相,至液层在分液活塞中间。取萃取后水相40 mL稀释至合适的浓度,以钼酸铵分光光度法检测磷酸的含量。
1.6 聚合物负载铬氧化剂的合成
取10 g D301型弱碱树脂,加6 mol/L盐酸5 mL,浸泡0.5 h左右,不断搅拌,再加入5 g三氧化铬,再浸泡4 h以上,然后过滤,用少量去离子水洗涤,再依次用THF,乙醚洗涤,直至滤液无色为止,此湿树脂可直接进行氧化或在空气中风干。容量约为2.53 mmol Cr(Ⅵ)/g[8]。选用商品化的弱碱性阴离子交换树脂D301作载体,制得聚合物Cr(Ⅵ)氧化剂,生成季铵盐树脂的反应见式 (1):
P—CH2N(CH3)+H2O+CrO3→

(1)
式(1)中P表示阴离子交换树脂D301中聚合物大分子。
1.7 检测手段在氧化剂对PCl3氧化中的应用
在直径2 cm,长20 cm的带夹套的离子交换柱中加入9.71 g聚合物负载铬氧化剂,夹套中为高温循环水,对聚合物负载铬氧化剂先预热20 min,然后加入含0.5 mL PCl3的40 mL PCl3正己烷溶液,充分接触,冷却后收集流出的PCl3正己烷溶液,并用高纯水将未被氧化的PCl3和氧化生成的POCl3萃取到水中转化为亚磷酸和磷酸,使用钼酸铵分光光度法和氧化差减法分别检测其含量[9]。
2 实验结果与讨论
2.1 水中亚磷酸和磷酸检测手段的建立
2.1.1 钼酸铵分光光度法标准曲线
2.1.1.1 最大吸收波长
精确配制磷酸根浓度为0.8、1.6、2.4、3.2和6.4 μmol/L的溶液50 mL分别加入比色管中,加入5 mL混合发色剂,混匀;15 min后用1 cm比色皿在紫外分光光度计于600~1 100 nm范围内测定吸收光谱,结果如图1。
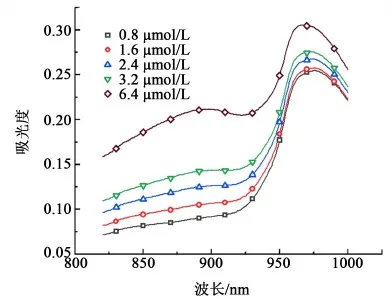
图1 磷钼蓝的吸收光谱Fig.1 Absorption spectrum of phosphomolybdic blue
从图1中可以看出,吸收峰出现在891和970 nm处,2处的扫描曲线相比较,891 nm处的曲线在低浓度时的分辨效果更好[10],且文献中选取的波长882 nm也与之相近,所以选择891 nm作为最大吸收波长。
2.1.1.2 标准曲线
精确吸取5价磷标准使用溶液0、0.10、0.25、0.50、1.00、2.00和4 mL于50 mL比色管中,每个样品均具有1个平行样,分别加入高纯水后稀释至50 mL;加入5 mL混合发色剂,混匀,15 min后,以0 mL的样品作为参比溶液,在891 nm处测定吸光度。
以吸光度为纵坐标,以比色管中磷酸浓度(μmol/L)为横坐标,进行标准曲线的绘制,结果如图2。
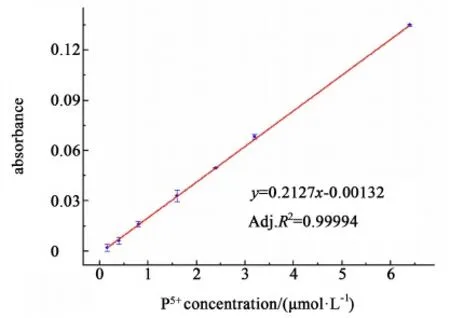
图2 钼酸铵分光光度法的标准曲线Fig.2 Standard curve of ammonium molybdate spectrophotometry
得到带误差棒的线性回归方程为:y=0.02127x-0.00132(Adj.R2=0.99994),具有可靠的线性拟合度。
分光光度法中,以扣除空白值后的吸光度为0.01 相对应的浓度值为检测限,代入线性回归方程,得出检测限为0.52 μmol/L,检测范围为0.52~6.40 μmol/L。
2.1.2 氧化差减法测水中3价磷含量工作曲线
2.1.2.1 氧化差减法可行性分析
钼酸铵分光光度法的原理是磷酸根和钼酸铵结合生成磷钼蓝,其在一定的波长下吸光度会与浓度呈正比;但磷钼蓝很容易被氧化生成黄色的磷钼杂多酸,从而失去检测效用。过硫酸钾经可行性实验验证其不影响磷酸根和钼酸铵结合生成磷钼蓝[11-13],并且其在120 ℃下会分解产生氧原子,对亚磷酸具有良好的氧化性。
分别取5 mL 5 价磷标准溶液加入到5个比色管中,室温下分别加入0、1、2、3和4 mL 过硫酸钾饱和溶液,均稀释至50 mL,加入5 mL混合发色剂,混匀,15 min后,在891 nm处测定吸光度,结果如图3。
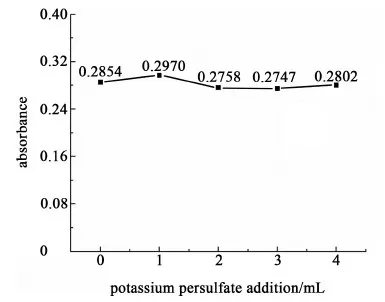
图3 过硫酸钾加入量对钼酸铵分光光度法的影响Fig.3 Effect of potassium persulfate addition on ammonium molybdate spectrophotometry
从图3中可以看出,在稳定的检测体系中饱和过硫酸钾溶液加入量不同,主体分子的吸光度较为稳定,变化不大,说明不加热至120 ℃的情况下过硫酸钾对于方法没有影响,不会出现将磷钼蓝氧化导致不能显色的情况。
2.1.2.2 氧化加热时间
取0.1 mL PCl3于烧杯中,加20 mL水反应,冷却后移至容量瓶中,稀释后亚磷酸溶液浓度为0.000 114 mol/L,分别取1 mL上述溶液,加入4个比色管中,其中1个比色管作为空白对照,其他3个比色管中加入3 mL 过硫酸钾饱和溶液,加水稀释至40 mL,密封好后分别120 ℃加热10、15和20 min,冷却后定容至50 mL, 加入5 mL混合发色剂,混匀,15 min后在891 nm处测定吸光度,结果如图4。

图4 过硫酸钾氧化时间选择Fig.4 Selection of oxidation time of potassium persulfate
从图4中可以看出,120 ℃下加热氧化10、15和20 min后的磷酸含量变化不大,氧化加热10 min后生成的磷酸的量即已经达到加入的3价磷的量,所以,选择加热时间为10 min。
2.1.2.3 氧化剂加入量
从图5中看出,加入1、2 和3 mL过硫酸钾饱和溶液氧化,吸光度比较稳定,均可将加入4 mL的3价磷标准溶液中磷全部氧化,过硫酸钾的量越少,越有利于检测的准确性,选择过硫酸钾饱和溶液的加入量为1 mL。

图5 过硫酸钾加入量选择Fig.5 Selection of potassium persulfate addition
2.1.2.4 氧化差减法的工作曲线
分别取0、1.0、1.5、2.0、3.0和4 mL 3价磷标准溶液加入到50 mL比色管中,以氧化差减法检测其中亚磷酸含量。
以差值吸光度为纵坐标,3价磷浓度(μmol/L)为横坐标,进行标准曲线的绘制,结果如图6。
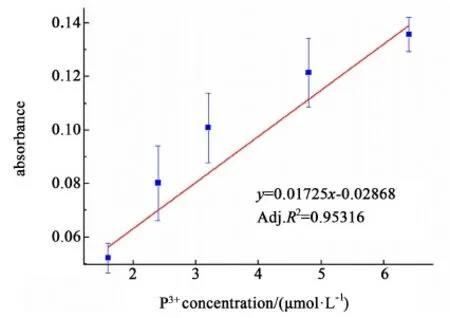
图6 氧化差减法的工作曲线Fig.6 Working curve of oxidation subtraction method
得到带误差棒的线性回归方程为:y=0.01725x+0.02868(Adj.R2=0.95316),线性拟合度较好,检测限为1.60 μmol/L,检测范围为1.60~6.40 μmol/L。氧化差减法得到水溶液中亚磷酸的浓度有良好的可靠性,以及这个方法可以用于检测水中亚磷酸的量。
2.2 探究不同条件对POCl3在正己烷-水萃取体系中萃取率的影响
采用水萃取并反应正己烷中磷元素的方法将其转移到水中,以磷酸或亚磷酸的形式存在,反应方程式见式(2)和(3):
POCl3+H2OH3PO4+HCl
(2)
PCl3+H2OH3PO3+HCl
(3)
已知有机溶液的体积为Vo,水溶液的体积为Vw,则萃取率E为:
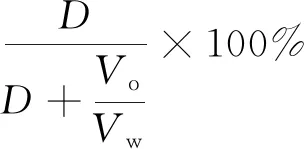
(4)

本研究考察了室温萃取20 min的条件下,相同体积比下不同起始POCl3浓度对萃取率的影响,以及相同初始浓度下不同体积比对萃取率的影响,结果如图7。

图7 体积比和POCl3浓度与萃取率的关系Fig.7 Effect of volume ratio and phosphorus oxychloride concentration on extraction rate
由图7中可以看出不同条件下萃取率与起始浓度和体积比的规律,以及拟合曲线。在其他条件相同的情况下,体积比与萃取率呈反比,起始浓度与萃取率呈正比。
由于起始浓度为0时萃取率为0,并且随着浓度增加,萃取率越来越接近于100,采用非线性拟合,选择y=a-bcx作为拟合公式,拟合结果见表1。

表1 不同体积比下磷起始浓度与萃取率的拟合方程Table 1 Fitting equation of phosphorus initial concentration and extraction rate at different volume ratios
根据拟合曲线以及萃取率与起始浓度和体积比的规律计算出不同体积比下目标浓度的萃取率,由于萃取率定义中Vo/Vw与萃取率倒数呈线性关系,可以使用拉格朗日插值法外推出目标体积比和起始浓度下的萃取率。
水溶液中磷酸的物质的量可以通过钼酸铵分光光度法测得,并测得实验条件下的萃取率,计算出正己烷溶液中的POCl3的量,这为萃取后使用钼酸铵分光光度计法计算正己烷溶液中5价磷含量提供了理论基础。
在室温下,分别向40 mL正己烷中加入0.000 4 mol的PCl3,溶解后用50 mL水萃取20 min,PCl3萃取后的水溶液使用氧化差减法测出其中亚磷酸的量为0.000 287 mol,得到萃取率与POCl3在相同条件下的萃取率相同为70%;多组实验的结果可以说明,PCl3和POCl3在相同的条件下萃取率相同。
2.3 聚合物负载铬氧化剂
聚合物负载铬氧化剂的容量约为每1 g 2.53 mol Cr(Ⅵ),该氧化剂几乎不溶于所有试剂,并且具有良好的热稳定性,可以95 ℃下保持稳定[10]。聚合物负载铬氧化剂氧化PCl3的反应在油浴的密封烧瓶中和循环水夹套固定床中进行,PCl3被氧化剂氧化会生成POCl3,而聚合物中的Cr(Ⅵ)被还原,以含氧酸根离子的形式存在于聚合物D301树脂中。
2.4 聚合物负载铬氧化剂固定床氧化效果检测
聚合物氧化剂被装载于固定床之后,在循环水温度下预热45 min后,降低循环水温度,物料通过固定床,对PCl3的氧化效果如表2所示。
65 ℃的条件下,取30 min后流出的1 mL检测,氧化率低于1%; 95 ℃的条件下,取30 min后流出的1 mL检测,以100 mL高纯水室温下萃取20 min,对萃取后的水样中亚磷酸和磷酸含量进行检测。
当萃取率为100%,测得出正己烷溶液中PCl3和POCl3初始浓度均为32.5 mmol/L,拟合后萃取率结果如表3所示。

表2 聚合物负载铬氧化剂对PCl3的固定床氧化结果Table 2 Mixed bed oxidation results of PCl3 by polymer-supported chromium oxidant

表3 32.5 mmol/L磷初始浓度在不同体积比下的萃取率Table 3 Extraction rate of phosphorus at an initial concentration of 32.5 mmol/L at different volume ratios
根据萃取率定义使用拉格朗日插值法外推出体积比为0.01时的萃取率为99.93%,计算出氧化后正己烷溶液中实际的PCl3和POCl3含量,经多次平行实验结果得出转化为POCl3的氧化率约为25.6%。
对一次实验中固定床氧化中95 ℃被还原的聚合物负载铬氧化剂、负载后的聚合物负载铬氧化剂以及未负载的聚合物D301树脂进行了X射线衍射实验,结果如图8所示。

图8 被还原后聚合物负载铬氧化剂,聚合物负载铬氧化剂,以及未负载的聚合物D301树脂的XRD谱图Fig.8 XRD pattern of chromium (Ⅵ)-supported polymer after reduction, chromium (Ⅵ)-supported polymer, and unsupported polymer-D301 resin
图8中可以看到21.4°处在负载后有明显的特征峰,但在氧化后可以看到21.4°的峰有明显的降低;首先可以说明CrO3被成功负载在D301树脂上;其次可以得出在氧化后有大量的6价Cr在反应后被还原,侧面说明聚合物负载铬氧化剂有良好的氧化效果,与钼酸铵分光光度法的检测结果相符合。
3 结论
为了建立准确及时地判断出溶剂中的磷含量的检测手段,本研究建立了钼酸铵分光光度法和氧化差减法测定水中亚磷酸和磷酸含量的检测手段,探究了POCl3和PCl3在正己烷-水萃取体系中不同条件下萃取率的影响规律,实现了对正己烷溶液中POCl3和PCl3的检测;最后采用检测手段展示了聚合物负载铬氧化剂对于PCl3的氧化效果,得到以下结论。
1)钼酸铵分光光度法和氧化差减法可以同时测定水中的磷酸和亚磷酸含量,并且得到吸光度与浓度均呈线性关系,磷酸的检测范围在0.52~6.40 μmol/L,检测限为0.52 μmol/L;亚磷酸的检测范围为1.60~6.40 μmol/L,检测限为1.60 μmol/L;并使用正己烷-水萃取体系萃取率规律实现检测正己烷溶液中POCl3和PCl3的含量。
2)聚合物负载铬氧化剂可以将PCl3氧化成为POCl3,在直径2 cm,长20 cm固定床中流速0.24 mL/min温度95 ℃的条件下,多次实验结果表明对PCl3转化生成POCl3的转化率可以达到26.5%。
