三嗪超交联聚苯乙烯基多孔聚合物的设计与制备
2020-12-09邱玉倩刘千惠韩浩杰王洪强
邱玉倩, 刘千惠, 韩浩杰, 于 涛, 王洪强, 徐 飞
(1. 西北工业大学材料学院,凝固技术国家重点实验室,纳米能源材料研究中心,西安 710072;2. 西北工业大学柔性电子研究院,西安 710129)
多孔聚合物具有孔结构丰富、比表面积大、密度低以及功能化设计灵活等特点,在气体吸附、分离、催化、传感、储能等领域引起了关注[1-9]。其中,超交联多孔聚合物是一类通过超交联反应(如傅克烷基化反应)得到高度交联的多孔聚合物,具有原料易得、合成简单等优势,展现出广泛的应用前景[10-16]。根据原料不同,超交联反应主要分为两类:(1)以有机小分子作为原料直接进行聚合与超交联[3];(2)以聚合物作为原料进行超交联反应[17,18]。前者通常得到超交联微孔聚合物[19],而后者可得到具有微孔、介孔或大孔的层次孔聚合物[20];同时,以聚合物为原料亦可通过其结构(如形貌)设计来进一步调控超交联多孔聚合物的微观形貌及孔结构[17,21,22]。因此,以聚合物为原料制备超交联多孔聚合物引起了愈来愈多的关注。
采用聚苯乙烯为原料制备超交联多孔聚合物,一般包括如下步骤:首先将聚苯乙烯溶解或者充分溶胀(对于预交联聚合物),随后选择合适的温度和催化剂使溶解或溶胀的聚合物发生超交联反应[3]。最早的超交联多孔聚合物由Tsyurupa和Davankov[23,24]在上世纪70年代以聚苯乙烯为原料经傅克烷基化反应制备得到。目前,常用的交联剂包括二甲氧基甲烷[2,12]、1,2-二氯乙烷[2]和四氯化碳[20]等,以其构筑亚甲基或者羰基的交联桥结构。交联桥结构对所制备超交联多孔聚合物的微观形貌、孔结构与功能位点等有很大的影响,进而决定着超交联聚合物或其衍生材料在吸附、分离与催化等方面的性能。因此,开发新型交联结构对于超交联多孔聚合物的结构设计以及性能优化具有重要意义。
本文以三聚氯氰作为交联剂,通过傅克烷基化反应在聚苯乙烯中构建三嗪交联桥结构,获得了三嗪超交联的聚苯乙烯基多孔材料。研究表明,利用线性聚苯乙烯为原料制备的三嗪超交联多孔聚苯乙烯由较小的纳米颗粒堆叠,形成三维纳米网络层次孔结构;其中颗粒内部含有交联形成的微孔,颗粒间堆叠形成丰富的介孔。与羰基超交联制备的多孔聚苯乙烯相比,三嗪超交联的聚苯乙烯具有更大的比表面积(最大可达945 m2/g)和更丰富的介孔结构。同时,引入的含氮三嗪环功能位点将在电化学储能、催化等性能方面具有潜在的优势[25-27]。此外,采用预交联的聚苯乙烯球为原料时,也可以获得三嗪超交联的微米或亚微米聚苯乙烯球,球内含有丰富的微孔。在上述基础上,进一步研究了聚合物原料浓度与超交联时间对所制备材料孔结构的影响。本文提出的三嗪交联策略有助于丰富和拓展超交联多孔聚合物结构,在催化、吸附缓释和储能等领域具有潜在的应用前景。
1 实验部分
1.1 原料和试剂
无水乙醇:分析纯,西陇化工股份有限公司;十二烷基苯磺酸钠:分析纯,天津市科密欧化学试剂有限公司;二氯甲烷、四氯化碳、丙酮、乙醇、过硫酸钾、苯乙烯、无水氯化钙:分析纯,国药集团化学试剂有限公司;三聚氯氰(CC)、二乙烯基苯、无水氯化铝:分析纯,上海阿拉丁生化科技股份有限公司;线性聚苯乙烯颗粒(LPS):广州金发科技有限公司;商用聚苯乙烯泡沫:上海佳奈包装材料公司。
1.2 测试与表征
采用美国FEI公司Nova NanoSEM 450型场发射扫描电子显微镜(SEM)以及FEI Talos F200X型透射电子显微镜(TEM)表征样品的微观形貌;采用美国Thermo Fisher Scientific公司Nicolet iS50傅里叶红外光谱仪(FT-IR)测试分析材料的化学结构;采用美国Micromeritics公司ASAP2020型吸附仪测定样品的氮气吸附-脱附等温线,称取约70 mg样品,在110 ℃真空下脱气干燥6 h后进行测试;采用Brunauer-Emmett-Teller(BET)方法计算所测样品的比表面积(SBET);用单点法计算样品吸附孔隙的总孔隙体积(VTOTAL);用t-plot法计算样品的微孔表面积(SMIC)、微孔体积(VMIC)和外部孔表面积(SEXT);以非定域密度泛函(DFT)模型计算孔径分布。
1.3 实验步骤
1.3.1 预交联聚苯乙烯球(SPS)的制备 将150 mL溶剂(超纯水或乙醇)和150 mg表面活性剂(十二烷基苯磺酸钠)加入三颈烧瓶中,随后加入3.75 mL苯乙烯和0.375 mL二乙烯基苯,在氮气保护气氛下搅拌5 min(搅拌速率为250 r/min),物料分散均匀后,再加入150 mg过硫酸钾。将体系加热升温至75 ℃并且搅拌反应3 h后,再次加入0.375 mL二乙烯基苯,并在75 ℃氮气保护气氛下继续搅拌反应24 h。反应体系降到室温后,将得到的白色乳状液体在10 000 r/min的转速下离心分离,去除上清液后加入超纯水再次洗涤并离心分离,重复上述操作3次。将所得白色沉淀放入60 ℃恒温干燥箱中干燥24 h,即得到SPS,样品标记为SPS-d,其中d为SPS的直径。样品SPS-295 nm和SPS-2.64 μm制备过程中的溶剂分别为超纯水和乙醇。
1.3.2 三嗪超交联聚苯乙烯基多孔聚合物的制备 取预定量的LPS加入50 mL二氯甲烷中,室温搅拌溶解或溶胀3 h后,再加入0.74 g CC和1.5 g无水氯化铝后加热至50 ℃冷凝回流反应20 h。待降温后,抽滤并用二氯甲烷洗涤。随后,将固体产物加入到50 mL、1 mol/L的盐酸-丙酮(体积比为1∶ 3)混合溶液中搅拌3 h后抽滤,重复以上操作2~3次。最后,在60 ℃恒温干燥箱中干燥24 h得到超交联产物,命名为LPS-CC-x,其中x为加入LPS 的质量,x为0.2、0.6、1.5、2.0 g。
当采用SPS为原料进行超交联反应时,步骤与LPS-CC的制备类似,SPS的用量为0.6 g,产物命名为SPSCC-m-n,其中m为SPS的直径,n为超交联反应时间,分别为5 min、1 h、2 h、10 h和33 h。
1.3.3 羰基超交联聚苯乙烯基多孔聚合物的制备 取0.2 g LPS加入50 mL四氯化碳中,搅拌溶解3 h后加入1.5 g无水三氯化铝,并将温度升至75 ℃反应24 h。待反应液冷却到室温后抽滤,并用四氯化碳洗涤。将所得固体转移至50 mL、1 mol/L的盐酸-丙酮(体积比为1∶3)混合液中搅拌3 h后抽滤,重复上述操作2~3次。最后,在60 ℃恒温干燥箱中干燥24 h后得到羰基超交联产物,命名为LPS-CO-0.2 g。
2 结果与讨论
2.1 三嗪超交联线性聚苯乙烯基多孔聚合物的制备及结构表征

图1 以三聚氯氰作为交联剂制备三嗪超交联多孔聚苯乙烯的示意图Fig. 1 Schematic diagram of preparation of triazine hypercrosslinked porous polystyrene
通过傅克烷基化反应在聚苯乙烯中构筑新型的三嗪交联桥结构,可获得三嗪超交联的聚苯乙烯基多孔材料(图1)。首先,将LPS溶解在二氯甲烷中,再以CC为交联剂、三氯化铝为路易斯酸催化剂,经傅克烷基化反应,使CC脱去氯原子带正电后,与LPS上的苯环发生交联可获得三嗪超交联结构的聚苯乙烯多孔材料[28-30]。超交联反应后样品由LPS的白色转变为LPS-CC-0.2 g的棕黄色(图2(a、b)),且LPS-CC-0.2 g与反应前相比,质量增加48%。这些现象初步表明发生了超交联反应。进一步从红外光谱(图2(c))可以看出,LPS-CC-0.2 g除在1 450、1 600 cm-1处出现与LPS类似的苯环骨架伸缩振动峰和吸收振动峰外,也在1 512、1 385 cm-1处出现了新的三嗪基伸缩振动峰,证明了三嗪交联结构的存在[28]。

图2 LPS (a) 交联前和 (b) 交联后的照片; (c) 样品的红外光谱图Fig. 2 Digital images of LPS (a) before and (b) after hyper-crosslinking; (c) FT-IR spectra of samples
超交联后样品的微观形貌如图3(a~c)所示,LPS-CC-0.2 g是由粒径为十几纳米的不规则颗粒相互三维堆叠形成的含有介孔和部分大孔的三维纳米网络结构(图3(a)和(b))。与此同时,从高分辨TEM可以看到,颗粒内部存在大量微孔(图3(c))。氮气吸附-脱附测试进一步分析了LPS-CC-0.2 g的孔结构(图3(d)),在低压区等温曲线急剧上升,表明LPS-CC-0.2 g具有微孔结构;在中高压区出现明显的滞后回环,证明其存在丰富的介孔,在相对压力接近1时,吸附曲线缓慢上升,表明LPS-CC-0.2 g内部存在少量大孔。上述结论也在DFT孔径分布图(图3(e))中得到证实。LPS-CC-0.2 g的孔径在微孔、介孔和大孔区域均有分布,属于层次孔聚合物,通过BET模型计算其样品比表面积高达945 m2/g。
三嗪交联桥结构与羰基交联桥结构相比,其产物孔结构差异显著。通过氮气吸附-脱附测试得到,LPSCC-0.2 g的比表面积明显高于LPS-CO-0.2 g的相应值(表1),且样品孔径集中分布在4~10 nm。而LPS-CO-0.2 g的孔径在微孔、介孔和大孔区域都有一定的分布,孔径分布较宽。可见相较于羰基交联体系,本文的三嗪交联体系构筑的超交联聚苯乙烯基多孔聚合物具有更大的比表面积和丰富的介孔,且孔径分布更为集中。
LPS加入量对超交联反应产物的孔结构的影响见图4。在其他实验条件相同的情况下,通过调控LPS加入的质量,获得一系列LPS-CC样品。由表1可知,LPS-CC-0.2 g的比表面积最大,随着LPS质量的增加,所得产物比表面积有逐步降低的趋势。此外,研究表明,LPS浓度增加导致超交联产物的增重率从37%,34%降低到27%。不同LPS加入量所得产物的氮气吸附-脱附等温线如图4(a)所示。所有样品曲线均在低压吸附区急剧上升,说明样品含有微孔结构,且LPS加入量的变化没有对微孔产生明显影响。在中压区曲线出现明显的滞后回环,表明样品生成大量介孔。DFT孔径分布图(图4(b))和孔结构数据表明,随着LPS加入量的增加,2~10 nm孔径的介孔含量减少。此外,图4(a)曲线后半段高压区呈现缓慢上升的趋势并出现吸附平台,证明样品中的大孔较少。综上所述,在三嗪超交联反应中,一定范围内,LPS加入量越大,超交联样品的比表面积越小。其中LPS加入量对材料微孔结构影响较小,而介孔则随着LPS加入量的增加逐步减少。这主要由于交联反应过程中,聚苯乙烯溶解度逐渐下降,以表面能最低的颗粒析出,优先进行颗粒内交联形成微孔,而后颗粒之间继续发生交联形成介孔,LPS浓度主要对颗粒之间的交联影响较大。

图3 LPS-CC-0.2 g 的 (a) SEM、 (b) TEM、 (c) 高分辨 TEM 照片;LPS-CC-0.2 g 和 LPS-CO-0.2 g 的 (d) 氮气吸附-脱附等温线和 (e) DFT 孔径分布图Fig. 3 (a) SEM, (b) TEM and (c) high resolution TEM images of LPS-CC-0.2 g; (d) Nitrogen adsorption-desorption isotherms and (e) DFT pore size distribution curves of LPS-CC-0.2 g and LPS-CO-0.2 g

表1 LPS-CC的比表面积和孔结构参数Table 1 Specific surface area and pore volume of LPS-CC
图5(a、b)所示为预交联的聚苯乙烯球SPS-295 nm和SPS-2.64 μm的SEM照片,SPS呈现出优良的球形貌,粒径较为均一。采用 SPS 进行超交联得到的 SPS-CC-295 nm-33 h 和 SPS-CC-2.64 μm-33 h(图 5(c、d))样品亦呈现出均匀的单分散球形貌,其粒径分别为266 nm和2.63 μm(图5(e、f))。
2.2 三嗪超交联聚苯乙烯球基多孔聚合物的制备及结构表征
图6(a,b)分别为SPS-CC样品的氮气吸附-脱附曲线以及BFT孔径分布图。可以看出,交联时间极大地影响了样品的微孔结构,SPS-CC-295 nm-1 h样品的SMIC为31 m2/g,而SPS-CC-295 nm-2 h的SMIC则达到343 m2/g(图 6(c)),交联时间较长的样品(SPS-CC-295 nm-10 h,SPS-CC-295 nm-33 h)微孔含量则保持相对稳定。而上述样品的氮气吸附-脱附等温线在中压区均吸附量增加不明显,表明介/大孔在样品中几乎没有或数量极少,这是由于超交联主要发生在聚苯乙烯球的内部,导致球内微孔的发育,而球与球间则由于尺寸较大难以发生外部交联形成丰富的介孔/大孔。综上可知,随着反应时间的增加,聚苯乙烯球内部逐渐交联形成微孔结构,且该过程主要发生在交联反应的前2 h(图6(d))。

图4 LPS-CC 的(a) 氮气吸附-脱附等温线和 (b) DFT 孔径分布图Fig. 4 (a) Nitrogen adsorption-desorption isotherms and (b) DFT pore size distribution curves of LPS-CC

图5 (a) SPS-295 nm, (b) SPS-2.64 μm, (c) SPS-CC-295 nm-33 h 和 (d) SPS-CC-2.64 μm-33 h 的 SEM 照片; (e) SPS-CC-295 nm-33 h 和 (f) SPS-CC-2.64 μm-33 h 的粒径分布图Fig. 5 SEM images of (a) SPS-295 nm, (b) SPS-2.64 μm, (c) SPS-CC-295 nm-33 h, (d)SPS-CC-2.64 μm-33 h; Particle size distribution of(e) SPS-CC-295 nm-33 h and (f) SPS-CC-2.64 μm-33 h
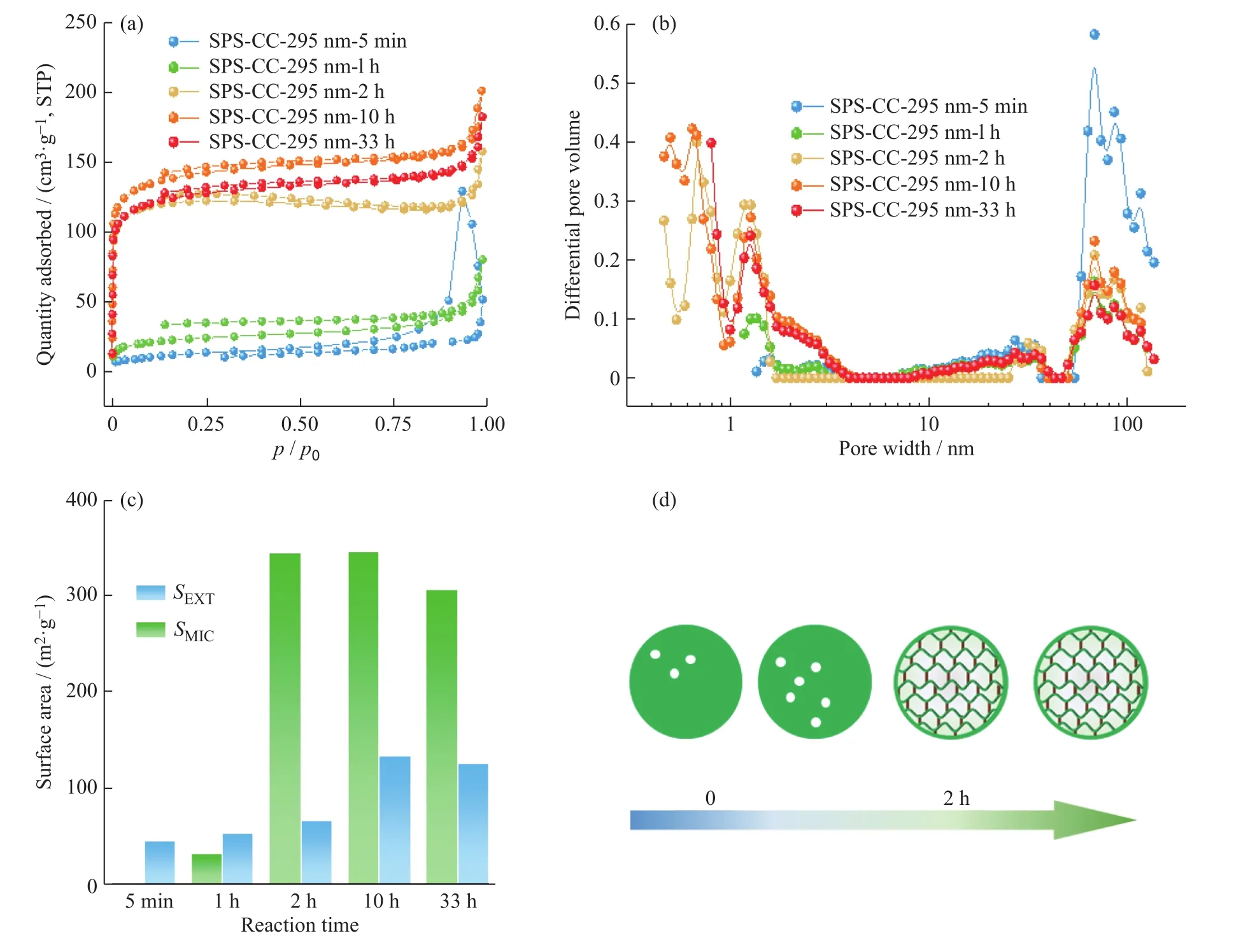
图6 SPS-CC-295 nm 的 (a) 氮气吸附-脱附等温线; (b) DFT 孔径分布图; (c) 不同交联反应时间下的比表面积(SEXT 与 SMIC);(d) 三嗪超交联多孔聚合物的示意图Fig. 6 (a) Nitrogen adsorption-desorption isotherms; (b) DFT pore size distribution curves; (c) Surface areas (SEXT and SMIC) of SPS-CC-295 nm at different reaction time; (d) Schematic diagram of triazine hypercrosslinking reaction
2.3 三嗪超交联聚苯乙烯基多孔聚合物的普适性
为了进一步研究三嗪超交联反应的普适性,将原料扩展到商用聚苯乙烯泡沫,将其进行相同的交联处理,也得到了具有相同类型层次孔结构的三嗪超交联聚苯乙烯基多孔聚合物,其比表面积高达1 101 m2/g,与传统的1,2-二氯乙烷作为溶剂及交联剂制备的超交联聚合物相比,比表面积约为后者的1.9倍(573 m2/g)[31]。目前,由于聚苯乙烯废弃后难降解,环境不友好[32,33]。因此,该交联策略也为对环境造成严重污染的大量聚苯乙烯废弃塑料的深加工及再利用提供了一个新思路。
3 结 论
(1)线性聚苯乙烯、聚苯乙烯球、商用聚苯乙烯均可以在三聚氯氰作为交联剂的条件下发生超交联反应构筑三嗪超交联聚苯乙烯基多孔聚合物,该交联剂具有一定的普适性。
(2)以LPS为原料时,通过控制LPS浓度,可以实现对超交联三维网络聚苯乙烯的介孔结构调控。随着LPS浓度增加,聚苯乙烯颗粒堆叠形成的介孔不断减少,材料比表面积和总孔容下降。
(3)以SPS作为原料时,超交联反应主要发生在SPS内部,从而形成单分散微孔球。随着超交联反应时间增加,球内微孔不断发育,在2 h内基本保持稳定。
(4)相同反应条件下,与羰基超交联材料相比,三嗪超交联的多孔聚合物具有更大的比表面积和更为丰富的4~10 nm介孔结构。
