右美托咪定对H9C2细胞内质网应激性损伤的影响及机制研究
2020-11-24朱志鹏凌晓燕张才军周清河周红梅
朱志鹏 凌晓燕 张才军 周清河 周红梅
内质网应激反应(endoplasmic reticulum stress,ERS)及细胞凋亡广泛参与了心肌缺血再灌注损伤(ischemia/reperfusion injury,IRI)的生理病理进程,通过对相关ERS信号通路以及作用靶点的调控发挥心肌保护作用[1-3],虽然人们通过细胞和动物实验不断探索可能的潜在机制,但目前具体作用机制仍不清楚。右美托咪定(dexmedetomidine,DEX)对重要脏器的IRI具有保护作用[4-6],且对缺氧/复氧(hypoxia/reoxygenation,H/R)性损伤的内皮细胞[7]及非IRI引起ERS信号通路的分子伴侣、蛋白和细胞凋亡呈现出积极作用[8]。为进一步明确DEX对心肌细胞损伤后ERS的影响及具体的信号通路,本研究通过构建H9C2心肌细胞H/R模型,探讨DEX对H/R下H9C2心肌细胞ERS的影响,并寻找可能的分子靶点。
1 材料和方法
1.1 细胞培养 H9C2大鼠胚胎心肌细胞株由嘉兴市第二医院实验中心提供,Annexin V/PI凋亡试剂盒 (上海索宝生物科技公司),CCK-8试剂盒(日本东仁化学科技有限公司,CK-04),逆转录试剂盒(美国Invitrogen公司),内质网应激特异性分子葡萄糖调节蛋白78(Glucose regulated protein 78,GRP78)、CCAAT/增强子结合蛋白同源蛋白(CCAAT/enhancer binding protein homologous protein,CHOP)、Caspase-12 特异性引物(上海索宝生物科技公司)。取出冻存的H9C2细胞,快速解冻,放入含有10%FBS和1%P/S的DMEM培养基(英国Gibco 公司,11995-065)中,以 1 000 r/min 离心 5 min,去上清液,细胞重悬,于37℃、5% CO2恒温细胞培养箱中培养,1~2 d换液,当细胞铺满整个培养皿底面积达到80%以上时,用0.25%胰蛋白酶(美国Gibco公司,16000-004)消化,并按1∶3进行传代或进行后续实验。将 DEX、4-苯基丁酸(4-phenylbutyric acid,4-PBA)和p38丝裂素活化蛋白激酶(p38MAPK)抑制剂(SB202190)均溶于二甲基亚砜(DMSO)溶液中,配置成各自目标浓度为1 mmol/L、100 μmol/L和5 mmol/L的原始储存液,分装后低温避光保存,短期使用前将其在水浴箱中迅速溶解,按1∶1 000比列加入培养基。
1.2 建立H/R实验模型验证DEX的干预作用 参照前期研究,设立缺氧3 h复氧3 h的心肌缺血模型。将对数生长期H9C2细胞培养至密度为70%左右,制作单细胞悬液。按细胞密度为3×105个/ml接种于96孔板,过夜,每组3个复孔,设立对照板和H/R板(即Control组、DEX组、H/R组、DEX+H/R组、4-PBA组、4-PBA+H/R组、4-PBA+DEX+H/R组)。其中对照板正常培养基常氧条件下培养,与各实验组同一时间点换培养基;H/R板换无血清无糖的DMEM培养液后放入含5% CO2和95% N2密闭缺氧装置(Billups-Rothenberg公司,MIC-101)中缺氧3 h,置换培养基常氧条件下培养3 h。DEX干预组(包括DEX组、DEX+H/R组和4-PBA+DEX+H/R组)在细胞贴壁后于H/R前1 h换溶解有1 μmol/L DEX的缺氧培养基,4-PBA干预组(包括4-PBA组、4-PBA+H/R组、4-PBA+DEX+H/R组)在H/R前的24 h加入培养基孵育,然后进行缺氧3 h复氧3 h的实验程序。收集细胞上清液采用低损伤法检测LDH浓度,CCK-8试剂盒检测细胞活力,流式细胞术检测细胞凋亡率,RT-PCR和 Western blot法检测细胞 GRP78、CHOP、Caspase-12 mRNA和蛋白的表达情况。
1.3 p38MAPK蛋白表达水平检测 采用毒胡萝卜素(TG组)体外孵育H9C2细胞10 h进行干预实验,DEX和4-PBA孵育同前。先将细胞分为对照(Control)组、H/R组和DEX+H/R组,实验结束收集各组细胞,采用Western blot法,以 β-actin为内参,检测 p38MAPK(稀释比 1∶1 000)和 p-p38MAPK(稀释比 1∶1 000)目的蛋白条带的灰度值,与内参条带灰度值的比值反映目的蛋白的相对表达水平。再选择SB202190进行信号通路关键分子分析,将孵育好的细胞随机分为对照组、TG组、DEX+TG组、DEX+TG+4-PBA组、SB202190+TG组、DEX+TG+SB2020190组、DEX+TG+4-PBA+SB202190组。其中,4-PBA预处理24 h,TG预处理10 h,DEX预处理1 h及SB202190预处理10 min,然后正常培养至所需时间。收集细胞,分别检测各组细胞活力、凋亡情况以及各组靶分子的基因和蛋白表达(检测内容与方法同1.2)。
1.4 细胞损伤检测 按照CCK-8试剂盒说明,设置空白对照组和观察组,每组设置3个复孔,于96孔板中每孔加入10 μl CCK-8,约1 h后上机酶标仪450 nm处测OD值,计算细胞活力(%)=(观察-空白)/(对照-空白)(空白组为只加入了培养基的阴性对照)。同时,根据各组不同实验时间点收集细胞培养上清液,根据LDH试剂盒要求,顺序加入检测试剂,酶标仪450 nm处检测OD值,计算细胞培养上清液中的LDH浓度。计算公式:细胞培养上清液LDH浓度=(测定OD值-对照OD值)/(标准OD值-空白OD值)×标准品浓度×N×1 000(N指稀释倍数)。
1.5 细胞凋亡检测 采用流式细胞AnnexinV/PI双染法,细胞以每孔2×105个铺6孔板,按不同的实验分组方案要求分组培养,收集细胞,并用预冷的PBS清洗2遍,每管细胞样品中加入500 μl Binding Buffer轻轻重悬细胞,加入 5 μl AnnexinV-FITC、5 μl Propidi Iodide,混匀,室温避光孵育15 min。上机检测,采用Sumol/lmit5.0软件进行分析。
1.6 RT-PCR检测mRNA表达 细胞经各种处理后,弃上清液,PBS冲洗细胞,按照RNA提取试剂盒的操作步骤说明,以Trizol法提取各组细胞总RNA,用逆转录剂盒将RNA逆转录为cDNA,分别取2 μl在PCR仪上配置25 μl反应体系进行扩增,以β-actin为内参,根据PCR反应条件:95℃预变性3 min,95℃变性30 s,55℃退火20 s,72℃延伸20 s,40个循环,72℃最后延伸 10 min。引物分别为:GRP78 上游:5′ACTGGAATCCCTCCTGCTC-3′,下游:5′CAAACTTCTCGGCGTCAT 3′;CHOP 上游:5′TGCCTTTCGCCTTTGAGAC-3′,下游:5′GCTTTGGGAGGTGCTTGTG-3′;Capsase-12 上游:5′GGGATAGCCACTGCTGATA-3′,下游:5′GCCACTCTTGCCTACCTTC-3′。计算CT值,采用相对定量(relative quantities,RQ)=2-ΔΔCt进行计算相对表达量。
1.7 Western blot检测蛋白表达 细胞经相应处理后,其充分裂解,冷却后离心取上清液,直接用于蛋白电泳上样。采用BCA法计算检测各组蛋白浓度,进行SDS-PAGE 电泳。分别以 CHOP(Affinity1∶1 000)、GRP78(Affinity1∶1 000)、Caspase-12(Affinity1∶1 000)一抗和辣根过氧化物酶(HRP)标记的羊抗小鼠二抗反应后,摇床上洗涤,进行化学发光反应,配置显影液,入化学发光成像系统进行化学发光,拍照。通过 ImageProPlus软件对发光条带进行定量分析。以β-actin为内参,各蛋白的相对表达量=蛋白电泳条带OD值/βactin产物电泳条带OD值。
1.8 统计学处理 采用SPSS 17.0统计软件、GraphPad Prism5.0作图软件。计量资料以±s表示,两组间比较采用独立样本t检验,多组间比较采用单因素方差分析,两两比较采用Dunnet或Tukey检验。P<0.05为差异有统计学意义。
2 结果
2.1 各组H9C2细胞活力、LDH浓度和细胞凋亡率的比较 与Control组相比,H/R组的细胞活力明显降低(P<0.05),LDH 浓度明显增加(P<0.05),细胞凋亡率增加(P<0.05);与H/R组相比,DEX+H/R组与4-PBA+H/R组的细胞活力明显增加,细胞上清液的LDH浓度降低,细胞凋亡率也降低,差异均有统计学意义(均P<0.05)。见表1和图1。
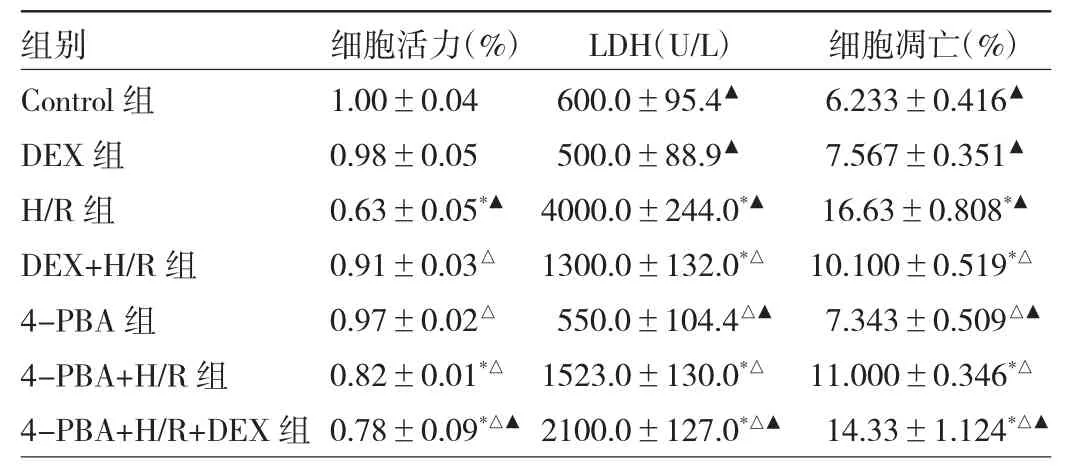
表1 各组H9C2细胞活力、LDH浓度和细胞凋亡率的比较
2.2 各组细胞GRP78、CHOP和Caspase-12 mRNA和蛋白表达的比较 与Control组相比,正常孵育下DEX组及4-PBA组在GRP78、CHOP和Caspase-12的mRNA和蛋白表达均无统计学差异(均P>0.05),而H/R组在3种基因和蛋白表达上分别一致上调(P<0.05)。与H/R组相比,DEX+H/R组与4-PBA+H/R组在GRP78、CHOP和Caspase-12的mRNA和蛋白表达均明显下调(均P<0.05)。与DEX+H/R组比较,DEX+H/R+4-PBA组的上述指标进一步反弹升高。见图2。
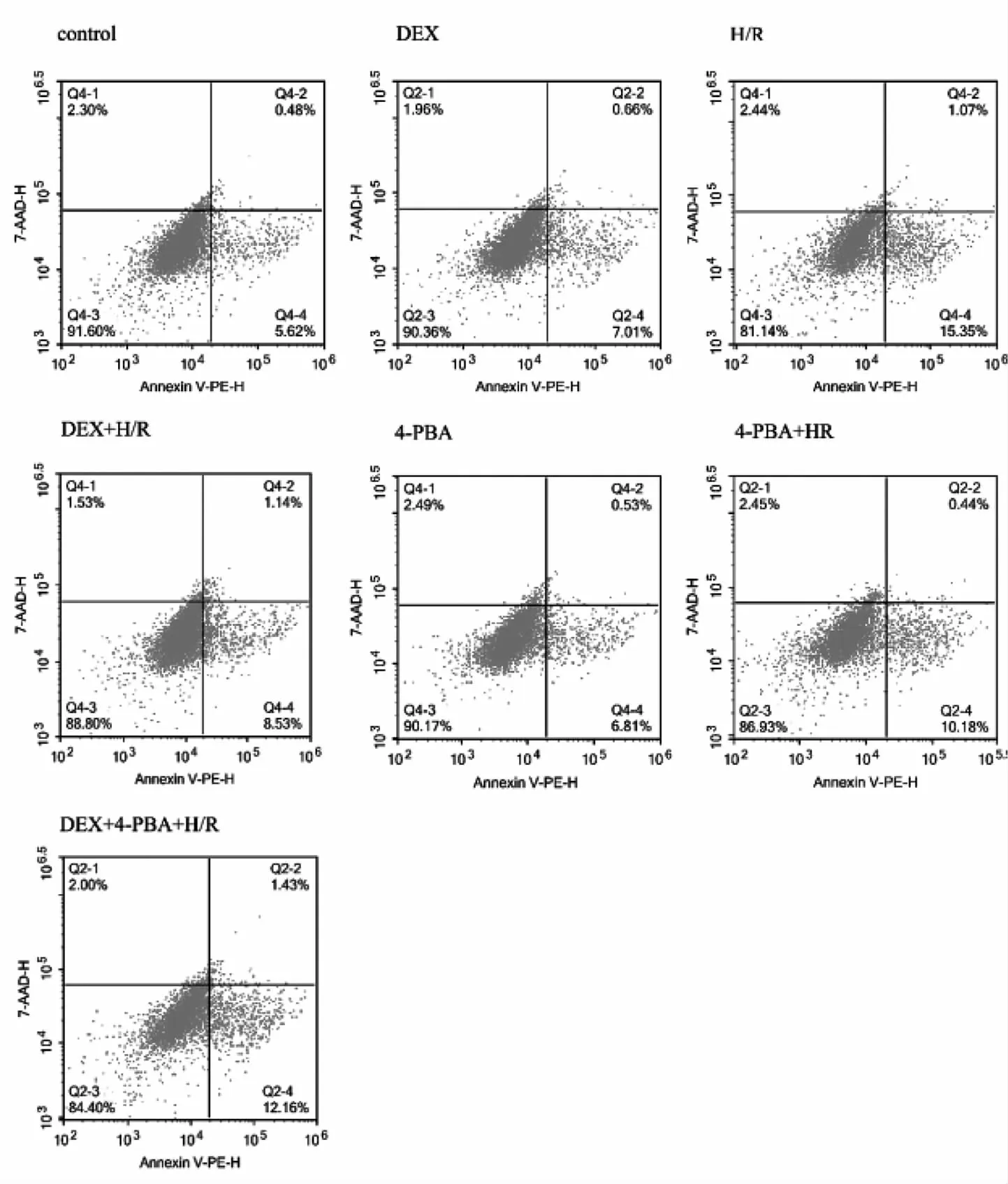
图1 各组细胞经相应处理后的流式细胞凋亡图(H为缺氧,R为复氧,DEX为右美托咪定,4-PBA为4-苯基丁酸钠盐)
2.3 各组细胞p38MAPK及p-p38MAPK表达及细胞活力、LDH浓度、细胞凋亡情况的比较 与Control组比较,TG组H9C2细胞p-p38MAPK蛋白表达水平明显升高(P<0.05),与 TG 组相比,DEX+TG 组 p-p38MAPK蛋白表达明显降低,差异有统计学意义(P<0.05)。而各组间p38MAPK蛋白表达水平差异无统计学意义(P>0.05),见表2。与Control组比较,TG组细胞活力明显下降,LDH浓度明显增高;与TG组相比,DEX+TG组的细胞活力增高明显,LDH浓度下降,细胞凋亡率明显改善(P<0.05);与 DEX+TG 组比较,SB202190+TG组的细胞活力、LDH浓度和细胞凋亡率均无统计学差异(均 P>0.05),DEX+SB202190+TG组的各项指标被p-p38MAPK抑制剂SB202190所逆转,呈现相反的方向变化(P<0.05),见表 3。
2.4 各组细胞不同处理后GRP78、CHOP、Caspase-12和p38MAPK mRNA和蛋白表达的比较 DEX+TG组及SB202190+TG组之间的GRP78、CHOP、Caspase-12 mRNA和蛋白表达均无统计学差异(均P>0.05);与TG组相比,DEX+TG组、DEX+TG+4-PBA组、SB202190+TG 组、DEX+TG+SB2020190组、DEX+TG+4-PBA+SB202190 组的 GRP78、CHOP、Caspase-12 mRNA 和蛋白的表达均降低(均P<0.05);与DEX+TG组相比,DEX+TG+4-PBA 组、SB202190+TG 组、DEX+TG+SB2020190组、DEX+TG+4-PBA+SB202190组均能够明显增加GRP78、CHOP、Caspase-12 mRNA 和蛋白表达的上调(均P<0.05),此外,DEX+TG+SB20219+4-PBA组的上调幅度最大。见图3。
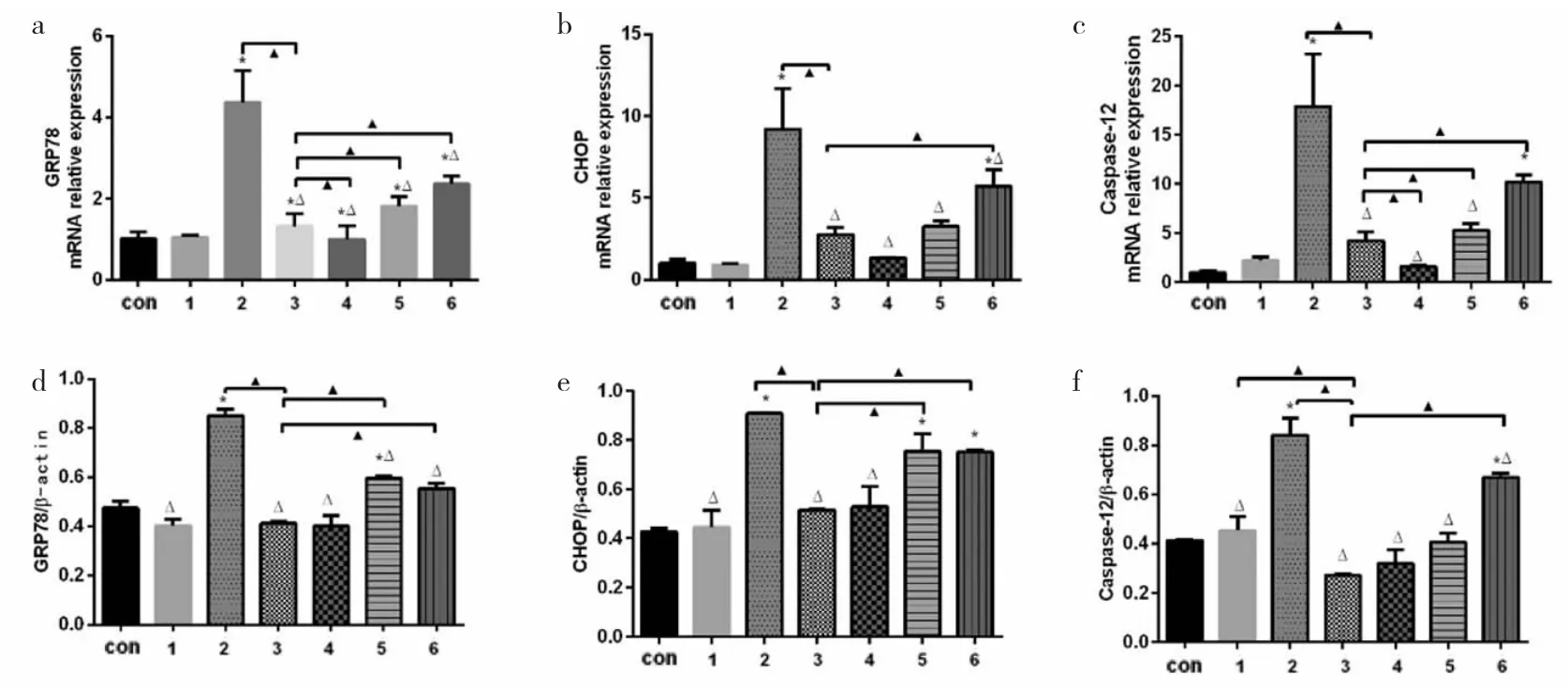
图2 各组细胞内质网应激特异性分子葡萄糖调节蛋白78(GRP78)、CCAAT/增强子结合蛋白同源蛋白(CHOP)、Caspase-12表达的比较(a、b、c分别为 GRP78、CHOP、Caspase-12 的 mRNA 表达差异;d、e、f分别为 GRP78、CHOP、Caspase-12的蛋白表达差异。Con为Control组,1为DEX组,2为H/R组,3为DEX+H/R组,4为4-PBA组,5为4-PBA+H/R组,6为4-PBA+DEX+H/R组。与Con组比较,*P<0.05;与 H/R 组比较,△P<0.05;与 DEX+H/R 组比较,▲P<0.05)
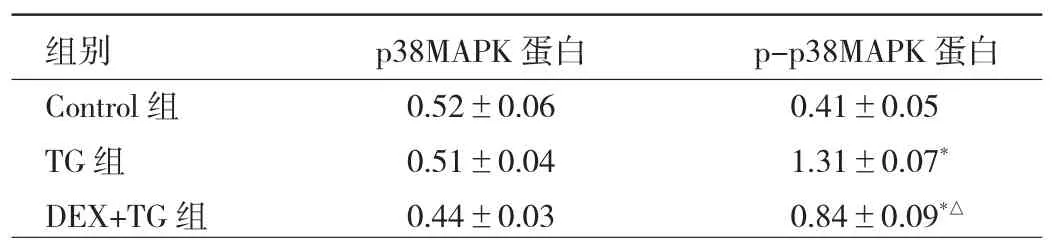
表2 DEX对TG诱导的H9C2细胞p38MAPK表达情况比较
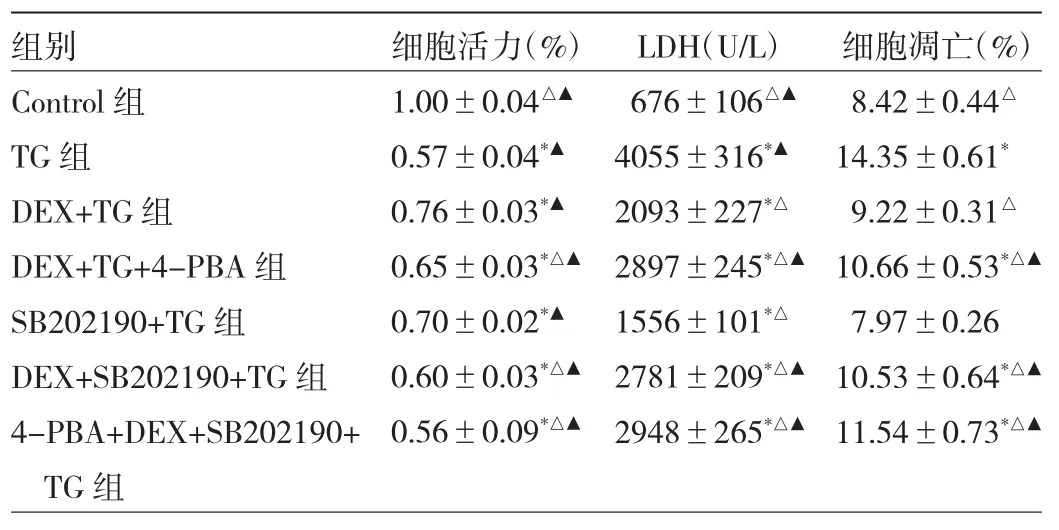
表3 DEX对H/R条件下的不同处理组H9C2细胞活力、LDH浓度和细胞凋亡情况的比较
3 讨论
心肌的缺血和再灌注损伤是临床常见现象,虽然多方位防治策略的应用在IRI基础研究中获得了成功,但是临床应用仍然难以改变患者结局及并发症[9]。近年来动物实验证实[10-12],DEX可以减轻心肌IRI后的ERS,调控心肌凋亡程序,具有心肌保护作用。本研究一方面采用前期实验的细胞模型,从心肌细胞内质网应激凋亡角度,研究DEX预处理对H9C2细胞H/R损伤的保护作用,另一方面运用TG诱导的H9C2内质网应激模型,探讨DEX对ERS产生保护机制的可能机制。
DEX是一种高选择性α2肾上腺素受体激动剂,近些年其器官保护作用吸引了大批学者的兴趣。缺血再灌注研究方面,DEX显示出具有一定的心肌保护[13]、肺保护[14]、中枢神经保护[15]和肾脏保护[16]作用,另外,DEX还具有炎症及相关信号通路调控[15,17]、细胞凋亡及自噬[18-19]等作用。ERS诱导的细胞凋亡是除了外源性凋亡途径和死亡受体凋亡之外的又一途径,是应激感知和调控凋亡信号的主要机制[20]。细胞在缺血缺氧后,引起ERS导致GRP78的高表达,同时启动未折叠蛋白反应稳定内质网功能,保持细胞稳态,主要通过ATF6、肌醇需酶和蛋白激酶R样内质网激酶(PERK)3条信号通路进行调控。当ERS反应失控以后,心肌细胞通过CHOP和Caspase-12信号通路的激活,而产生细胞凋亡。本研究结果证实右美托咪定可通过影响内质网应激,从而保护其对于H/R时H9C2细胞的损伤,最终下调H/R损伤细胞的GRP78、CHOP、Caspase-12表达,而利用内质网应激抑制剂4-PBA可以明显逆转DEX的保护效果,充分说明DEX是作用于ERS而发挥作用的。Peng等[21]利用原代乳鼠细胞H/R模型的研究显示,DEX也能够减轻心肌细胞H/R的损伤,只不过其实验模型设计为缺氧1 h复氧24 h。GAO等[17]对缺氧1 h复氧12 h的H9C2细胞进行DEX干预,发现1 μmol/L DEX能够明显减轻细胞损伤,与本实验采用的实验剂量及结果一致,但是模型参数不同,而且是通过白细胞介素的炎症通路来调控损伤。至于两者实验模型与本研究的差异,可能是来自于模型细胞的不同或目标研究通路来自于非内质网应激通路。
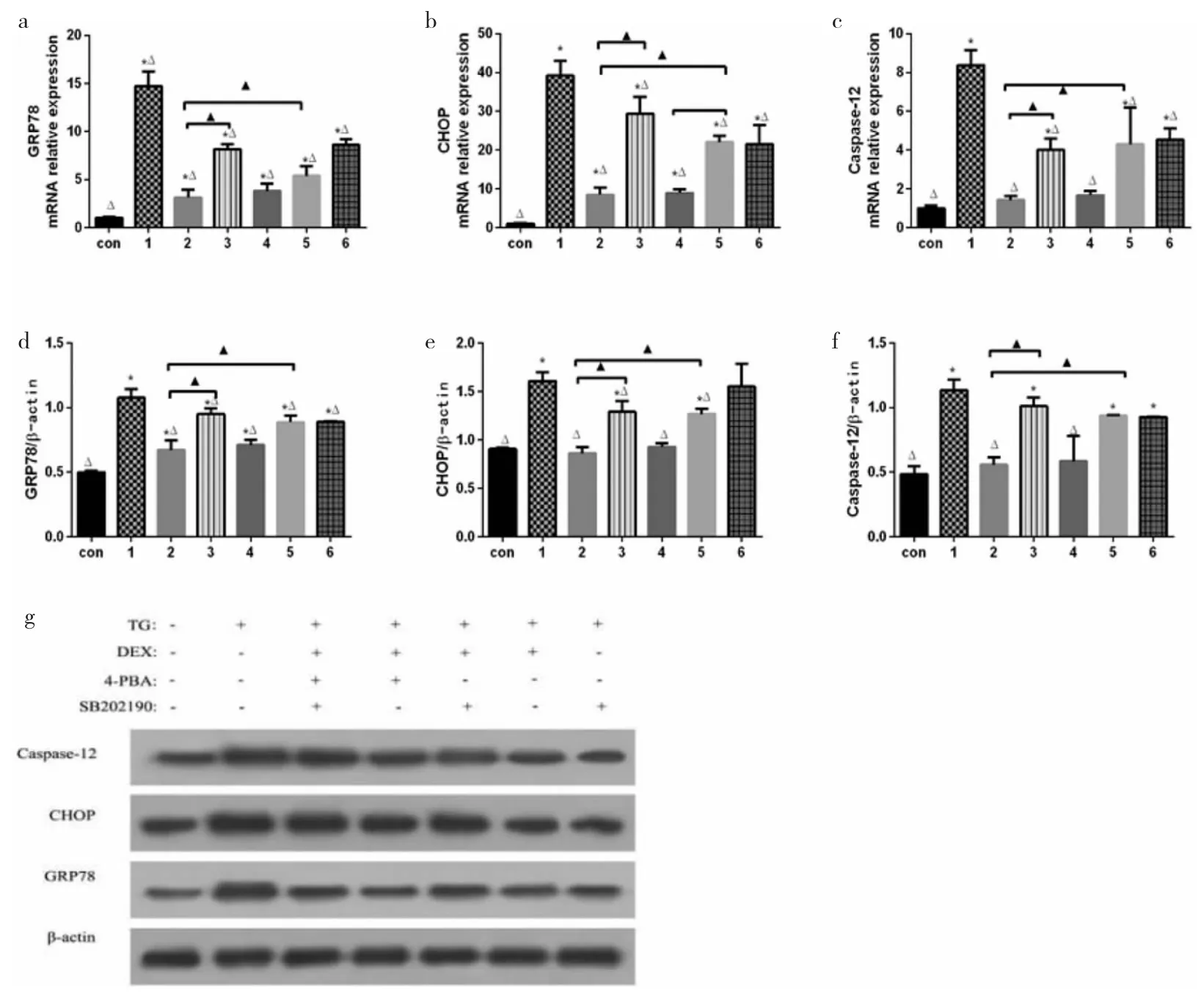
图3 各组细胞不同处理下内质网应激特异性分子葡萄糖调节蛋白78(GRP78)、CCAAT/增强子结合蛋白同源蛋白(CHOP)、Caspase-12 表达(a、b、c分别为 GRP78、CHOP、Caspase-12 的 mRNA 表达差异;d、e、f分别为 GRP78、CHOP、Caspase-12 的蛋白表达差异;g为GRP78、CHOP、Caspase-12蛋白电泳图。H为缺氧;R为复氧;DEX为右美托咪定;4-PBA为4-苯基丁酸钠盐;TG为毒胡萝卜素;SB202190为p38MAPK特异性抑制剂。其中,Con代表Control组;1代表TG组;2代表DEX+TG组;3代表DEX+TG+4-PBA组;4代表SB202190+TG组;5代表DEX+TG+SB202190组;6代表DEX+TG+SB202190+4-PBA组。与Control组比较,*P<0.05;与TG组比较,△P<0.05;与 DEX+TG 组比较,▲P<0.05)
p38MAPK是丝氨酸/苏氨酸激酶家族的成员,它是介导细胞反应的关键,通过上游丝裂原活化蛋白激酶(MAPK)磷酸化Thr-X-Tyr而被激活,在其众多家族成员中,p38MAPK与感染、炎症应激反应有关[22]。那么,p38MAPK是如何被激活的呢?首当其冲的原因就是ERS。研究显示,ASK1的自磷酸化可以导致MKK的活化和 p38MAPK 的激活[23];Cardoso 等[24]研究指出,ERS可以激活多重信号通路,例如eIF2α、MAPK和NF-κB,从而引发下游级联反应,MAPK进一步激活MAPK3/MAPK6,产生p-p38MAPK。除此之外,感染和其他原因引起的ERS也能够诱导p38MAPK的激活[25]。本研究中TG诱导的ERS产生了与上述同样的实验结果,p-p38 MAPK的表达明显增加。
那么,在经典的p38MAPK信号通路中,p-p38MAPK会导致什么下游变化呢?据报道,p-p38MAPK能够导致组织细胞 NF-κB 介导的炎症[26-27]、自噬[28-29]和凋亡[30-31]等进程。例如,雍陟等[32]研究发现,在心肌缺血再灌注细胞中,p38MAPK通路明显激活,结果导致心肌细胞的凋亡明显增加,Sanit等[33]通过抑制p38MAPK信号通路,减轻缺血再灌注损伤。本研究中,笔者首次采用DEX预处理,得到了相似的实验结果,即内质网应激产生了p-p38 MAPK的高表达,通过激活p38MAPK信号通路,最终导致心肌凋亡的增加,而DEX预处理能够抑制p38MAPK的激活,从而发挥类似于4-PBA的ERS凋亡保护作用。至于p38MAPK激活与下游凋亡信号通路的关系研究,不在本研究范围之内,有待进一步研究。
综上所述,H9C2细胞H/R时会导致异常的ERS和凋亡,伴随p38MAPK信号通路的激活,DEX可通过干预内质网应激,调控p38MAPK以及下游凋亡信号通路,发挥心肌保护作用。
