醇类有机物热传导的分子动力学模拟及微观机理研究
2020-11-18刘万强杨帆袁华张远达易平贵周虎
刘万强,杨帆,袁华,张远达,易平贵,周虎
(湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,湖南省高校QSAR/QSPR重点实验室,功能膜材料湖南省工程研究中心,湖南湘潭411201)
引 言
传热是化工生产领域最基本的问题之一,而热导率(thermal conductivity, λ)是表征物质导热性能的一个重要物性参数,λ 越大,导热性能越好。根据热导率的数值,可以推算出导热介质所传导或者交换的热量。因此,热导率是化工、石油和能源等工程领域需要的基础数据。因此有机物热导率的准确测量和计算具有非常重要的理论和实用价值[1-3]。醇类有机物是最常见的一类有机物,对醇类化合物的传热过程和热导率的研究不仅具有实际意义,而且可以为其他有机物的传热研究提供参考。
然而目前热导率的实验测量耗时较长并且对设备要求高[4]。因此研究者们提出了一些估算有机物热导率的方法。一类是从经验发展起来的估算公式,但该方法适用的有机物结构和种类有限且计算误差较大,并且在计算时常常需要该物质的其他性质的实验数据[5-7]。另一类方法基于定量构效关系(quantitative structure-property relationship,QSPR)模型,这种方法计算误差较小,但必须先建立分子参数和热导率之间的定量模型才能使用,难以满足实际需要[8-9]。
近年来,分子动力学(molecular dynamics,MD)模拟方法应用于液体的热传导以及微观过程研究已取得一些进展[2,10-17]。表1 列出了近年来采用MD 方法对醇类有机物导热过程的模拟和热导率的计算结果。可以发现:(1)研究的化合物的种类较少;(2)通过傅里叶定律可以得知,温度会对醇类有机物的热传导产生影响,但目前的研究没有考察温度对有机物导热的影响;(3)对于液体导热的微观机理的研究虽取得一定进展,但仍然需要进一步深入研究[11-12]。
使用MD 方法对有机物热传导过程进行模拟,优点在于能够获得实验中难以探测的微观信息,例如原子水平的能量转移和途径等[10],便于获得原子间的相互作用[22-23]、化学结构[24]和振动模式[25]等对热传导的贡献。因此本文采用非平衡分子动力学(non-equilibrium molecular dynamics,NEMD)方法初步探索了分子结构和温度对醇类有机物传热的影响,模拟了8 种醇类有机物在4 种不同温度下的热传导过程,通过对模拟过程数据的分析,初步探究不同微观热传导模式之间的关系和醇类有机物热传导的微观机理。

表1 部分醇类有机物的分子动力学模拟研究结果Table 1 Results of some MD simulation for calculating thermal conductivity of alcohols
1 理论基础和模拟方法
1.1 分子模拟的构建
对一系列醇类有机物(从乙醇到正壬醇共8 种)的热传导过程进行了分子动力学模拟研究。模拟体系的初始构象采用Materials Studio 软件中的Amorphous Cell 模块构建。由于PCFF 力场(polymer consistent force field)适用于聚合物和有机物的分子动力学模拟[26-28],因此采用PCFF 力场建立醇类有机物的分子模型。PCFF 力场相互作用的势能主要分为键合能(bonded energy)和非键合能(non-bonded energy)两类。其中,键合能包含键伸缩(bond stretching)、键 角弯 曲(angle bending)、二面 角扭 曲(torsion angle)、离面角作用力(inversion angle)和上述四项的交叉耦合项(combination cross)。非键合能包含分子间的库仑(Coulomb) 作用项和9-6 Lennard-Jones势能项(其截断距离为12.5 Å,1 Å=0.1 nm)。这样共有6类作用项。

图1 非平衡分子动力学模拟模型示意图Fig.1 Schematic diagram of NEMD simulation model
1.2 NEMD模拟模型、原理和过程
本文采用NEMD[29-30]方法对醇的热传导过程进行模拟,其模拟模型如图1所示。
当系统平衡后,热源(heat source)中粒子的热能(thermal energy)将有规律地增加,即热源粒子的速度将依照式(1)变化:

式中,vs是热源粒子s 的速度;vc是热源粒子的质心速度;Δε为常数,等于热源中能量的增量;Erk是粒子相对于其在热源中的质心的动能。
同样地,冷源(heat sink)中的粒子s 的速度将按照式(2)有规律地减小:

每隔一定的时间间隔Δt,热源和冷源的粒子将进行一次速度交换。根据冷源和热源粒子之间速度能量转换,可计算得到热通量J(heat flux),如式(3)所示:

式中,A 是模拟系统垂直于热能传递方向的截断面积。经历一段时间的冷热源粒子的速度交换,即可建立稳定的温度梯度,此时可通过傅里叶定律(Fourier’s law)计算得到热导率。如式(4)所示:

式中,dT/dz代表z方向的温度梯度;负号表示热能传递方向与温度梯度方向相反。
基于Algaer 等[2]的研究发现,对于一个在传热方向上有几纳米长,并且含有上千个分子的分子液体模拟体系,模拟盒子尺寸大小对模拟结果的影响并不明显。因此,本文不探究不同尺寸和模拟结果的相关性,并且将模拟盒子在x、y 和z 方向的初始长度分别设置为27、27 和216 Å。为了得到模拟系统的温度梯度分布,模拟盒子沿z 方向被分为40 层。同时为了避免界面效应(surface effect),在x、y 和z 方向都添加了周期性边界条件(periodic boundary condition)。具体模拟过程及优化后的参数如下。
(1)使用Brown 等[31-37]针对热通量计算修正后的LAMMPS(large-scale atomic/molecular massively parallel simulator)软件进行分子动力学模拟。在模拟过程中系统的势能和粒子的受力通过PCFF 力场计算得到。
(2)在弛豫前,对模拟体系进行结构预优化,避免不合理的原子间距离和受力,使体系的能量最小化。
(3)为了使体系在给定温度下平衡稳定,首先分别在NPT、NVT 和NVE 系综下进行了140 ps(200000步)的动力学弛豫来对体系进行预平衡。然后再分别在NVT 和NVE 系综下分别进行700 ps(1000000步)的动力学弛豫。
(4)当体系弛豫平衡后,继续在NVE 系综下,根据非对称eHEX (enhanced heat exchange)算法[38],进行总共2 ns 的热传导模拟过程,并在模拟体系的z方向添加恒定热流,其大小为Δε=0.003 kcal‧mol-1‧fs-1(1 cal=4.18 J)。
(5) 在弛豫过程中,根据Velocity-Verlet 方法进行数值迭代。
(6)由于模拟体系中H 原子的最小振动周期为10 fs,因此积分步长(time step)设置为0.7 fs。
(7)模拟过程在一个大气压下进行,并且温度分别设置为273、288、298 和323 K。模拟体系的温度和压力分别由Nose-Hoover[39]热浴法(thermostat)和压浴法(barostat)进行调控。
(8) 体系的长程静电相互作用(Long-ranged electrostatic interaction)根据Ewald法进行计算得到。
(9)模拟过程中当热导率和温度梯度收敛或稳定后,输出模拟结果并分析数据。
1.3 热流分解
根据不同的相互作用类型,由冷源和热源之间动能交换得到的总热通量Jtot,可分解为相应部分热通量,并主要分为传输项(transport term,Jtrans)和相互作用项(interaction term,Jinteract)两大类[18,23-24]。

传输项Jtrans主要包含由模拟体系中粒子迁移传递的能量,可分为动能项(kinetic energy term,Jkin)和势能项(potential energy term,Jpot):

式中,Us、ms和vs分别代表粒子s的势能、质量和速度;VCV代表冷源和热源之间的过渡区域(control volume),求和表示过渡区域中粒子动能或势能的总和。
相互作用项Jinteract主要由粒子间通过相互作用项进行能量传输。相互作用项包含非键合作用部分的分子间相互作用项(intermolecular interaction,Jinter)和键合部分的分子内相互作用项(intramolecular interaction,Jintra)。分子间相互作用项Jinter可分为范德华相互作用项JvdW和库仑相互作用(Coulomb interaction)项JCl。分子内相互作用项Jintra包含了四个相互作用项:Jstr、Jang、Jtor和Jinv,分别对应于键伸缩(bond stretching)、角 弯 曲(angle bending)、二 面 角(torsion angle)和离面角(inversion angle)。在液体中,相互作用项一般占主要部分[18,24]。

式中,fX代表作用于粒子s 的某一作用类型X 的相互作用。
根据式(5)~式(7)的分析,总的热通量Jtot可以分解为:动能项Jkin、势能项Jpot、范德华力项JvdW、库仑力JCl、键伸缩Jstr、角弯曲Jang、二面角Jtor和离面角Jinv共8个部分。根据不同类型的热通量JX,热导率也可被分 为 8 类 部 分 热 导 率 (partial thermal conductivity,λX)。

除超过一定距离的粒子间相互作用对外,每一种相互作用力都可视为一条粒子间传热路径(atomistic heat path)[24],而热能沿这个路径进行传输。每一种相互作用力X 的对应的部分热导率λX等于传热路径密度(heat path density,ρX)与传热效率(efficiency,ΛX)的乘积。

其中,传热效率指单位时间内通过单位面积热能传递速率。对于分子内相互作用项(X=键伸缩、角弯曲、二面角和离面角),每一个多体相互作用可视为一条传热路径,其密度为:

2 结果与讨论
2.1 热导率的计算
本文采用NEMD 模拟了8 种液态醇类有机物在273、288、298、323 K 下的热传导。表2 列出了根据式(3)、式(4)计算得到的32个热导率计算值与实验值的比较。从表2 可以看出,热导率的计算值与实验值的平均相对偏差为3.77%,最大相对偏差为4.94%,计算值与实验值基本吻合,表明采用的NEMD 模拟方法及优化的参数用于模拟计算醇类有机物的热导率较为理想。
从表2 可以看出,虽然热导率的实验值与计算值之间存在偏差,但本文的热导率计算值与实验值的变化趋势一致。另一方面,热导率的计算值和实验值的偏差,说明模拟时存在部分偏差如质量密度和力场等仍需改进。根据质量密度的模拟值与实验值(附录表A1)的比较可知,最终得到的模拟值低于实验值,但是两者随链长和温度的变化趋势相同。本文使用的PCFF 力场属于全原子(all-atom)力场,分子中某些自由度(包含氢原子的键伸缩项、角弯曲项等)在实际中属于基态(ground states)振动模型,而在模拟中做柔性键伸缩项或角弯曲项处理。因此在模拟中这类键伸缩项或角弯曲项可用来传递能量,并使热导率的计算值与实验值产生偏差。虽然上述误差使热导率的计算存在不确定性,但本文对分子传热机理与分子链长和温度的关系的研究仍然具有意义。

表2 NEMD模拟计算得到的热导率与实验值[1]比较Table 2 Comparison of thermal conductivity of experimental values and simulated values of alcohols obtained by NEMD simulation
2.2 热流分解
根据式(5)~式(7)对热通量进行了分解,得到了不同相互作用类型的热通量数据(数据请见附录表A2)。
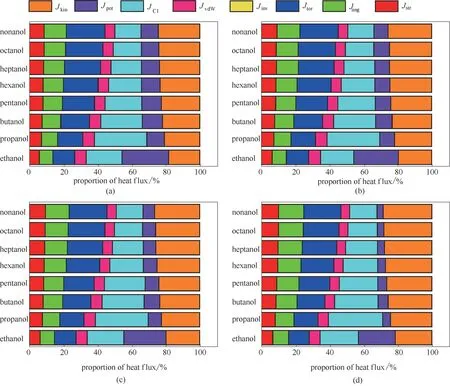
图2 273 K(a)、288 K(b)、298 K(c)和323 K(d)8种醇的不同作用类型相互作用热通量相对大小Fig.2 The proportion of heat flux of alcohols at 273 K(a),288 K(b),298 K(c)and 323 K(d)respectively
图2为273、288、298和323 K时8种醇的部分热通量相对大小示意图。从图2 可以看出:①动能项Jkin、二面角项Jtor和库仑作用项JCl在其中所占比例较大。这说明在醇类分子中主要通过分子的运动、分子内成键原子间振动和分子间库仑作用传热。②以图2(a)为例,273 K 下从乙醇到壬醇,动能项Jkin所占的比例从18.2%逐渐增大到24.4%,但势能项Jpot变化并不明显。由于分子链的增长,使得分子间碰撞的概率增加,因此动能项Jkin所占比例增加较显著。③分子内相互作用键伸缩Jstr,键角弯曲Jang,二面角Jtor的热通量所占比例随分子链的增长而增大。其中二面角项Jtor所占的比例最大且增加最为显著。273 K 下三者贡献值的总和从26.6%升高到44.4%。④除乙醇外,随碳原子数目的增加,分子间库仑作用项JCl和范德华作用项JvdW所占比例逐渐减小,特别是库仑作用项JCl变化尤为显著。273 K 下两者所占比例之和从37.5%降低到21.2%。⑤对于乙醇分子,传输项中的势能项Jpot的热通量比例远大于其他醇类分子。乙醇与其他分子相比较,其分子极性更大,且易形成分子间氢键,分子间的作用更强,分子迁移的势能项更大,运动更剧烈,因而势能项Jpot的热通量比例更高。
根据上述分析发现,在醇类有机物中,当分子链的碳原子数为5~6 时,热通量的主要部分将由分子间相互作用项Jinter转为分子内相互作用项Jintra。图3为己醇在不同温度时部分热通量的变化。

图3 不同温度下己醇的各类相互作用热通量比较Fig.3 Comparison of heat flux of different interactions of hexanol at different temperatures
从图3 可以看出:①在传输项中,动能项Jkin热通量最大且随着温度的升高显著增大(314.87~363.52 MW·m-2),可见由于温度升高,分子的整体运动速度加快,因此动能项的热通量显著增加。但是模拟体系随着温度升高密度减小(具体数值见附录表A1),分子间距离增大,势能项Jpot急剧减小(149.18~63.02 MW·m-2)。②随着温度升高,范德华作用项JvdW减小(85.98~72.88 MW·m-2),库仑相互作用 项JCl略 有 增 加(262.54~272.83 MW·m-2)。 在PCFF 力场中,库仑作用势能反比于原子之间距离r,而范德华作用为9-6 Lennard-Jones 势,与(1/r12-1/r6)呈比例关系。因此,随着温度的升高,原子间距离增大,范德华作用项与库伦相互作用均减小,但库仑作用项的变化较小。③在分子内相互作用项中,随着温度的升高,原子的振动加剧,导致键伸缩和键角弯曲项作用力增大,使得键伸缩项Jstr(111.14~128.15 MW·m-2)和键角弯曲项Jang(161.58~183.90 MW·m-2)都增加,二面角项Jtor减小(272.23~258.53 MW·m-2)。
综上所述,随着分子链增长,对热流的贡献由分子间相互作用项Jinter转为分子内相互作用项Jintra,这与Matsubara 等[18,24]和Ohara 等[40]的研究结果基本一致。其中,二面角项Jtor和动能项Jkin所占比例随分子链增长而增加,并占主导地位。此外,随着温度的升高,分子的整体运动速度加快,模拟系统的质量密度降低,因此,在己醇中,传输项的热通量Jtrans(包含Jpot、Jkin)随温度升高而升高,而相互作用项Jinteract(包含Jstr、Jang、Jtor、Jinv、JvdW、JCl)则降低(具体数值见附录表A2)。因此,最终的净效应是己醇的热导率随着温度的升高而降低。
2.3 原子传热路径分析
从2.2 节分析可以看出,随着分子链长的增加,分子内作用项Jintra显著增加并占主导地位。因而进一步考察了分子内各作用项的热导率(λX),并统计了模拟体系各项的密度(density, ρX),计算了各项的传热效率(efficiency,ΛX)。
图4(a)为273 K 下不同醇分子内键伸缩项、角弯曲项、二面角项和离面角项对应的部分热导率(partial thermal conductivity, λintra)。可以看出,键伸缩项部分热导率λstr、角弯曲项λang和二面角项λtor都随分子链的增长而增加。三者对整体热导率的贡献依次为λtor>λang>λstr。由于在链状分子中,几乎不存在离面角,因而λinv贡献几乎为零。此结论与烷烃的研究结果相似[24]。
图4(b)为273 K 下,模拟体系中键伸缩项、键角弯曲项、二面角和离面角的密度(density,ρintra)随分子链长的变化。由于分子链增长,分子内共价键数目增加;此外,通过模拟体系质量密度的分析发现,分子链增长模拟体系质量密度也增加了(详见附录表A1)。因此如图4(b)所示,键伸缩项ρstr、角弯曲项ρang、二面角项ρtor和离面角项密度ρinv均随着分子链的增长而增大,其大小依次为ρtor>ρang>ρinv>ρstr。

图4 醇类有机物在273 K下各分子内相互作用项的部分热导率λintra(a),密度ρintra(b)和传热效率Λintra(c)Fig.4 The partial thermal conductivity λintra(a),the density ρintra(b),and the efficiency Λintra(c)of intramolecular interaction paths for alcohols at 273 K,respectively
图4(c)为273 K 下,不同醇分子内键伸缩项、键角弯曲项、二面角和离面角作用项的传热效率(efficiency,Λintra)。由于模拟体系质量密度随分子链增长而增大,原子间碰撞概率增加,使得单位时间内通过单位面积的热能传递速率增大。因此,从图中可以看出,除了乙醇分子外,键伸缩项Λstr、键角弯曲项Λang和二面角项Λtor的传热效率都随分子链的增长而增大。此结果与Matsubara 等[24]对液态烷烃的研究结果相似。此外,当分子链较短时,如乙醇,分子的柔性较小,二面角的作用更强,因此二面角项Λtor较大。但当分子链增长时,二面角项Λtor逐渐减小,最后与键伸缩项的Λstr几乎相等,键角弯曲项Λang次之。
3 结 论
本文采用NEMD 模拟方法,模拟了8 种常见醇类有机物在不同温度下的热传导过程。得到的热导率的计算值与实验值的相对平均偏差为3.77%,最大相对偏差小于5.00%。
通过热流分解、部分热导率和传热效率的分析,表明醇类有机物的分子结构对其热能传输有较大影响。当分子链较短时,对醇类热传导起主要作用的是分子的传输项(动能和势能项)及分子内的二面角项。随着分子链增长,分子内的共价键数目增多,热流的主要传导方式逐渐由分子间相互作用过渡到分子内相互作用项(键伸缩项、角弯曲项、二面角项)传导。
此外,温度对醇类有机物的热传导具有一定影响,随着温度的升高,与分子和原子运动和振动相关的作用项,如动能项、二面角项、键伸缩项等传导的热流增加,而与势能有关的作用项,如势能项、范德华力项传导的热流减小,最终使得醇类有机物的热导率随温度升高而减小。
本文对醇类有机物的分子动力学模拟研究和热导率的计算,为研究液体的热传导过程提供了微观依据,并为热导率的计算提供了新的途径,同时也有助于理解液体热传导的途径和方式。
