环氧氯丙烷与离子液体的交联过程研究
2020-11-18涂卓恒史名珍张效敏吴有庭胡兴邦
涂卓恒,史名珍,张效敏,吴有庭,胡兴邦
(南京大学化学化工学院,江苏南京210023)
引 言
二氧化碳(CO2)大量排放导致全球温室效应加剧,由此带来的一系列环境负面效应如海水酸化、极端天气等严重威胁到人类的生存[1]。碳捕集、储存和利用(carbon capture, storage and utilization,CCSU)是近年来提出的用于解决CO2排放问题的关键技术之一,实现CO2的高效捕集则是最为重要的一步[2-3]。以单乙醇胺(MEA)为代表的有机胺吸收法是工业上应用最为广泛和成熟的CO2捕集技术,然而该技术仍存在着高操作能耗、高腐蚀性、低溶剂稳定性等诸多缺陷[4]。膜分离技术是近年来新兴的一类绿色分离方法,相比于传统的吸收法,其具有低能耗、低成本、模块化、连续化操作等优势。遗憾的是,目前以聚合物膜为代表的膜材料的CO2分离性能仍然受Robeson 上限制约,无法同时实现很高的CO2渗透系数和选择性[5]。
离子液体是一类具有极低挥发性、不可燃性、可设计性、酸性气体亲和性等优良性质的绿色溶剂,在CO2[6-11]、SO2[12-16]、H2S[17-19]、CO[20-22]等气体的吸收捕集领域展现出无可比拟的优势。将室温离子液体(RTILs)引入商业化聚合物膜制备离子液体支撑液膜(SILMs)是一种有效的改性策略,然而由于CO2在RTILs 中的渗透行为遵循最基本的溶解-扩散机理,故该类支撑液膜中CO2的渗透系数大多低于1000 barrers(1 barrer=7.52×10-18m3(STP)·m-2·m·s-1·Pa-1),CO2/N2理想选择性很少超过40[23-25]。为了进一步提高CO2分离性能,研究者们进一步以功能化离子液体(TSILs)制备支撑液膜用于CO2的选择性分离,如胺基功能化离子液体[26]、二胺质子型离子液体(PILs)[27]、氨基酸功能化离子液体[28]、苯酚功能化离子液体[29]、二羧酸功能化离子液体[30]等离子液体支撑液膜,同时实现了较高的CO2渗透系数(>2000 barrers)和选择性(CO2/N2选择性>100, CO2/CH4选择性>50)。
虽然离子液体支撑液膜在选择性分离CO2方面已表现出令人满意的性能[31],但其耐压性能依然饱受诟病(一般不超过2 bar, 1 bar=105Pa)。研究者们提出将离子液体固相化的策略以提高离子液体膜的耐压性能,如制备聚离子液体膜[32-34]、离子液体-聚合物混合膜[35]等。离子液体凝胶膜是一类易制备的高性能材料,可应用于CO2分离[36-37]、电化学[38]等领域,其中不可逆化学凝胶具备良好的热稳定性,在制备气体分离膜方面更具优势。McDanel 等[39]以多胺作为单体,双环氧基咪唑类离子液体作为交联剂制备了一类环氧-胺离子凝胶膜。Dai等[40]报道了以聚乙二醇二环氧甘油醚(PEGDE)作为交联剂,多胺离子液体或醚基二胺作为单体,聚乙二醇二甲醚(PEGDME)作为添加剂制备离子凝胶膜。目前来看,该类环氧-胺离子液体凝胶膜仅在较高添加剂(RTILs 或聚乙二醇衍生物)用量条件下展现出较为理想的CO2分离性能,其仍存在交联剂成本高或制备步骤复杂等问题。因此,未来的研究重点仍应集中在筛选廉价高效的交联剂,探究新的交联方法及交联机理,以期制备具有更高CO2分离性能和稳定性的离子液体凝胶膜。
针对上述问题,本文提出以二胺(或多胺)质子型离子液体为单体,以低成本、高活性的环氧氯丙烷为交联剂,制备具有高CO2分离性能的交联质子型离子液体(CPILs)凝胶(图1)。为了探究该设想的可能性,本工作探究了环氧氯丙烷与一种典型的二胺有机酸盐离子液体([DMAPAH][MOAc])在乙醇-水溶剂体系中的交联动力学,包括与酸中和顺序、反应温度、交联比例对环氧基团和氯基团的转化率和反应速度的影响。同时使用傅里叶变换红外光谱(FT-IR)和电喷雾质谱(ESI-MS)证明反应机理,使用在线监测黏度变化的方法测定该类环氧氯丙烷-二胺交联产物的凝胶转变性能。此外,初步测试了该类CPILs中CO2的渗透数据,探究交联反应对分离性能的影响。

图1 二胺质子型离子液体及环氧氯丙烷结构Fig.1 Chemical structures of DMAPA-based PIL and crosslinking agent used in this work
1 实验材料和方法
1.1 材料
CO2和N2气体(纯度为99.99%(mol))采购于南京创达气体有限公司;3-二甲胺基丙胺(DMAPA,分析纯)、甲氧基乙酸(MOAc,分析纯)购于Adamas试剂有限公司;环氧氯丙烷(分析纯)、乙醇(分析纯)、乙二醇(分析纯)、硫酸(分析纯)、盐酸(分析纯)、硝酸钾(分析纯)、氯化钾(分析纯)、丙酮(分析纯)均购于国药试剂有限公司。pH 标准缓冲溶液(pH 为6.86、9.18)购于上海仪电科学仪器有限公司。亲水性PES 平板膜(平均厚度100 μm,孔隙率80%,有效膜面积64 cm2)购于北京升河诚信膜有限公司。
1.2 仪器设备
(1)pH 监测:雷磁PXSJ-216型离子计,配备雷磁E-201-C 型pH 复合电极及雷磁T-818-B-6 温度探头。
(2)pX 测定:雷磁PXSJ-226 型离子计2 台(分别用于氯离子及环氧浓度测定),配备雷磁PCl-1-01型氯离子选择电极、雷磁C(K2SO4)-1 参比电极以及雷磁T-820D温度探头。
(3)表征仪器:FT-IR 谱图由Nicolet iS 10 型傅里叶红外变换光谱仪测定,ESI-MS 谱图由LCQ Fleet型电喷雾质谱仪测定,黏度由Brookfield LVDV-II+Pro型黏度仪测定。
1.3 交联反应动力学测试方法与步骤
主要通过pH 电极和氯离子电极监测反应体系中pH、氯离子浓度([Cl-])和环氧浓度([EPO])(盐酸-丙酮法)的变化,来获得交联反应动力学信息,同时考察与酸中和顺序、反应温度、交联比例等实验条件对交联反应的影响。具体实验装置、所需溶液及实验步骤如下。
1.3.1 电极活化及校正 使用pH 为6.86 和9.18 的标准缓冲溶液在对应温度条件下通过两点校正法校正pH电极。将氯离子选择电极在10-3mol/L NaCl溶液中浸泡活化,2 h后取出与参比电极一起用去离子水清洗至电位为-110 mV左右。之后使用浓度为1×10-3、3×10-3、1×10-2、3×10-2mol/L 的氯离子标准缓冲溶液在对应温度条件下通过四点校正法校正氯离子选择电极。为保证标准溶液与待测样品化学环境相同,用于氯离子浓度测定的氯离子标准溶液中含5%(体积)的0.15 mol/L的硫酸水溶液、1%(体积)的0.520 mol/L 的DMAPA(或[DMAPAH][MOAc])乙醇-水溶液(乙醇含量25%(质量),下同)以及1%(体积)饱和硝酸钾溶液(作为离子强度调节剂);用于环氧浓度测定的氯离子标准溶液含6%(体积)的0.3 mol/L 的盐酸丙酮溶液、1%(体积)的0.520 mol/L 的DMAPA(或[DMAPAH][MOAc])乙醇-水溶液以及1%(体积)饱和硝酸钾溶液。
1.3.2 交联反应动力学测试实验步骤 预先准备21 个25 ml 的具塞锥形瓶,10 个锥形瓶中加入5 ml 0.15 mol/L 的硫酸水溶液,另外11 个锥形瓶加入6 ml 盐酸丙酮溶液,密封备用。反应开始前取样1 ml 迅速加入装有6 ml 盐酸丙酮溶液的锥形瓶中,密封并摇匀,置于暗处3 h,加入1 ml 离子强度调节剂,用去离子水稀释并转移至100 ml 容量瓶,定容至刻度,用以测量盐酸丙酮溶液中氯离子浓度。
准确移取0.0520 mol/L的DMAPA(或[DMAPAH][MOAc])乙醇-水溶液(使用乙醇-水混合溶剂是为了保证环氧氯丙烷完全溶解,反应在均相状态下进行)100.00 ml 加入250 ml 三颈瓶中,加入磁子搅拌,插入pH 电极、温度探头,密封并在所需实验温度下恒温10 min。称取对应摩尔比的环氧氯丙烷加入三颈瓶,密封并开始计时,分别在第2、5、10、15、20、30、45、60、90、120 min记录pH。
同时在每个时间点取样两次,每次1 ml,第一次取出的样品迅速加入装有5 ml 0.15 mol/L 硫酸水溶液的锥形瓶中淬灭反应,密封并摇匀,静置5 min,加入1 ml 离子强度调节剂,用去离子水稀释并转移至100 ml 容量瓶,定容至刻度,混匀后取50 ml 溶液加入100 ml 烧杯,将洗净擦干的氯离子选择电极、参比电极和温度探头放入溶液中,待读数稳定后记录所测pX1值,该时间点反应体系中的氯离子浓度[Cl-](mol/L)计算公式为式(1):
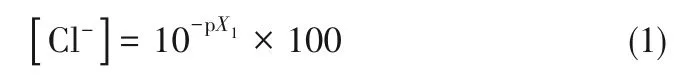
根据每个时间点测得的氯离子浓度计算转化率并对时间作图即得到[Cl-]-t曲线。使用滴定法检验该测试方法的准确性,发现两种方法的测试结果相对误差在1%以内。
第二次取出的样品迅速加入装有6 ml 盐酸丙酮溶液的锥形瓶中,密封并摇匀,置于暗处3 h,加入1 ml 离子强度调节剂,用去离子水稀释并转移至100 ml容量瓶,定容至刻度,用以测量环氧基团转化率随时间变化曲线。
取反应开始前处理配制用以测量盐酸丙酮溶液氯离子浓度的溶液50 ml 加入100 ml 烧杯,将洗净擦干的氯离子选择电极、参比电极和温度探头放入溶液中,待读数稳定后记录所测值pX0。取第二次取样处理配制的溶液50 ml 加入100 ml 烧杯,将洗净擦干的氯离子选择电极、参比电极和温度探头放入溶液中,待读数稳定后记录所测值pX2,该时间点反应体系中的环氧基团浓度[EPO](mol/L)计算公式为式(2):

根据每个时间点测得的环氧基团浓度计算转化率并对时间作图即得到[EPO]-t曲线。
1.4 凝胶转变性能研究
凝胶转变性能是环氧氯丙烷交联的二胺有机酸盐质子型离子液体的重要性质之一。考虑到溶剂的挥发对黏度仪的损伤以及测试数据偏大的问题,以乙二醇作为反应溶剂(1.2 ml),ECH 与DMAPA为反应物(摩尔比1∶1,共1 ml),在不同温度条件下测定反应体系的黏度随时间变化曲线,观察是否有凝胶点出现。
1.5 气体分离性能测试
交联质子型离子液体支撑液膜的制备及气体的渗透性能测试方法与作者之前工作[27,29]一致,此处不再赘述。
2 实验结果与讨论
2.1 交联动力学实验结果
2.1.1 合成方法及交联比例的影响 交联质子型离子液体的合成方法分为先中和后交联和先交联后中和两种(图2),首先采用先中和再交联的方法,以[DMAPAH][MOAc]作为交联反应单体,其与环氧氯丙烷1∶1 交联过程中反应体系pH 随时间变化曲线和氯基团转化率随时间变化曲线如图3所示。从图3(a)可以看出,由于[DMAPAH][MOAc]上一个胺基基团已被中和,所以体系的初始pH 较低,在25℃条件下反应两小时后,体系的pH 仅下降了0.4 左右,说明胺基与环氧氯丙烷反应较慢;从图3(b)可以发现,氯基团转化率也很低,在25℃条件下反应2 h 后仅有25%,即使温度提高至45℃,2 h 内氯基团的转化率只有60%左右。这主要由于DMAPA 被中和形成质子型离子液体后一个胺基被中和,DMAPA 亲核能力下降,胺基不能全部参与反应导致交联反应不能顺利进行。

图2 两种合成方法示意图Fig.2 Illustration of two synthetic methods used in this work
进一步改用先交联后中和的方法。在25℃条件下,DMAPA 与不同摩尔比ECH 交联的反应体系中pH、氯基团以及环氧转化率随时间的变化关系如图4所示。从pH随时间变化图中可以发现[图4(a)],由于反应单体为DMAPA,故体系具有更高的初始pH(12.2)。反应开始后,反应体系的pH 都随着时间的延长而快速下降。值得注意的是,当ECH 与DMAPA 摩尔比为0.2 和0.5 时,体系的pH 分别在30和45 min 后开始逐渐上升。氯基团优先与位阻更小的伯胺基团反应,生成弱酸性仲胺阳离子;仲胺阳离子可进一步与其他游离胺基交换质子,与氯基团进一步反应生成弱酸性的叔胺阳离子,上述步骤都是由动力学控制的。DMAPA 上的叔胺基团以及失去质子后的叔胺阳离子可与氯基团反应生成更稳定的中性季铵盐结构,这是热力学控制的。故反应开始阶段,弱酸性的仲胺、叔胺阳离子大量生成,体系碱性快速下降,而反应后期更多季铵盐结构的生成使体系的碱性有所回升。当ECH 与DMAPA 摩尔比为1.0 时,2 h 内体系的pH 从12.2 一直下降至10.3 左右,并没有出现回升的趋势。这主要是由于ECH 中和了更多的胺基,没有足够的游离胺基参与质子交换过程。综合上述,采用先交联再中和的方法更适合高交联度的交联离子液体制备。
从图4(b)、(c)中可以看出,相比于[DMAPAH][MOAc],以DMAPA 作为交联单体的反应体系氯基团转化速率较快,当ECH 与DMAPA 摩尔比分别为0.2 和0.5 时,2 h 内氯基团基本完全转化,环氧转化率也分别达到80%和50%左右;即使当ECH 与DMAPA 摩尔比提高至1.0时,2 h内氯基团也可转化90%左右,环氧基团转化率则相对较低,只有30%左右,这主要是由于氯基团和环氧基团与胺基的反应本质上是竞争关系,氯基团的反应活性高于环氧基团,一方面氯基团与胺基反应后产生的H+(或与叔胺形成季铵盐)将中和(或季铵化)胺基导致体系亲核能力下降,另一方面氯基团与DMAPA 反应后产物空间位阻增大,环氧与胺基的开环反应更难进行。

图3 不同温度条件下ECH与[DMAPAH][MOAc]等摩尔反应时体系的pH(a)和氯基团转化率(b)随时间的变化曲线Fig.3 Plots of pH(a)and conversion of chloride group(b)in the equimolar reaction system of ECH with[DMAPAH][MOAc]as a function of time at different temperatures

图4 25℃条件下ECH与DMAPA按不同摩尔比反应时体系的pH(a)、氯基团转化率(b)和环氧基团转化率(c)随时间的变化曲线Fig.4 Plots of pH(a),conversion of chloride group(b)and conversion of epoxy group(c)in the reaction systems with different ECH-to-DMAPA molar ratio as a function of time at 25℃
2.1.2 温度对反应的影响 为了进一步提高环氧基团的转化率,探究了ECH 与DMAPA 摩尔比1.0条件下反应温度对交联反应的影响,如图5 所示。从图5(a)中可以看出,当反应温度从25℃提高至55℃时,体系pH 下降更快且下降幅度更大,表明胺基的反应速率更快,与氯基团反应并被中和或季铵化的胺基基团数量也更多。从氯基团的转化率随时间的变化[图5(b)]中也可以看出,反应温度增加对提升反应速度有明显的作用,在反应温度为45℃和55℃条件下2 h 内氯基团基本完全转化。当反应温度由25℃提高至55℃时,环氧的转化率由25%左右提高至70%以上[图5(c)],这主要是由于一方面高温有利于胺基之间的质子交换进而释放更多游离伯胺和仲胺基团与环氧基团反应,另一方面高温也有利于胺基对环氧基团的亲核开环反应。考虑到反应温度从45℃提升至55℃以后氯基团及环氧基团转化率增幅很小,55℃是ECH 与DMAPA 交联的最佳温度条件。
2.2 凝胶转变性能

图5 不同温度条件下ECH与DMAPA等摩尔反应时体系的pH(a)、氯基团转化率(b)和环氧基团转化率(c)随时间的变化曲线Fig.5 Plots of pH(a),conversion of chloride group(b)and conversion of epoxy group(c)in the equimolar reaction systems of ECH with DMAPA as a function of time at different temperatures
为了探究ECH 与DMAPA 交联产物制备固膜或者凝胶膜的潜力,进一步测试了交联产物的凝胶转变性能。不同温度下乙二醇溶剂中ECH 与DMAPA(摩尔比1∶1)反应时体系的黏度随时间变化曲线如图6(a)所示。从图6(a)中可以发现,当反应温度为25℃和35℃时,3 h 内反应体系黏度增加极其缓慢(不超过1000 mPa·s),表明产物并没有交联为三维网络结构;而当反应温度为45℃和55℃时,3 h 内反应体系黏度急剧增加至4000 mPa·s 以上,表明该温度条件下体系逐渐转变为凝胶。由黏度的对数值对反应时间作图[图6(b)],根据曲线的拐点可判断体系的凝胶点。从图中可以看出,当反应温度为25℃和35℃时,体系没有出现明显的凝胶点,而当反应温度增加至45℃和55℃时,体系在140 和70 min 分别出现了明显的拐点,即为对应温度下体系的凝胶点。该结果进一步证明,ECH 与DMAPA 交联产物具有良好的凝胶转变性能,具备制备凝胶膜或者固膜的潜力。
2.3 交联反应机理
DMAPA 分子上的伯胺基团和叔胺可与环氧氯丙烷上的氯基团发生亲核取代反应;此外,伯胺基团可进攻环氧基团发生开环反应,生成的仲胺基团也可进一步与环氧基团发生反应。据此,提出了一种可能的交联机理,如图7所示。
进一步通过FT-IR 和ESI-MS 验证该机理。从图8 中可以看出,相比于未交联的DMAPA,交联产物的FT-IR 谱图(此处使用ECHn-DMAPA 而不是ECHn-[DMAPAH][MOAc]主要是为了避免[MOAc]-阴离子红外峰与部分特征峰重叠)出现新的红外峰。954 cm-1处的尖峰和3260 cm-1处的宽峰分别对应着新生成的—OH 的弯曲和伸缩振动,1129 cm-1处的峰归属于叔醇结构中的—C—O的伸缩振动,对应着环氧基团开环生成的异丙醇结构。同时,随着ECH与DMAPA 摩尔比的增加,C—N 键的弯曲振动(817~837 cm-1处的宽峰)逐渐减弱,说明胺基与环氧基团和氯基团发生了反应。此外,2533 cm-1处—NH2+伸缩振动表明铵离子的生成,说明氯基团与胺基发生了反应。

图6 ECH与DMAPA摩尔比1∶1时不同温度条件下反应体系黏度(a)、黏度对数(b)随时间变化曲线Fig.6 Plots of viscosities(a),logarithms of viscosity(b)in reaction systems with 1.0 of ECH-to-DMAPA molar ratio as a function of time at different temperatures
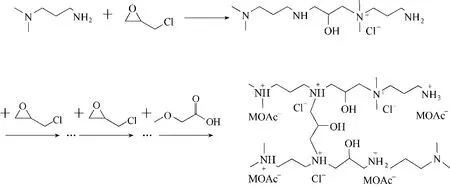
图7 环氧氯丙烷与[DMAPAH][MOAc]的一种可能交联机理Fig.7 One possible mechanism of ECH crosslinking with[DMAPAH][MOAc]

图8 不同ECH与DMAPA摩尔比的ECHn-DMAPA交联产物FT-IR谱图(反应温度55℃,反应时间2 h)Fig.8 FT-IR spectra of ECHn-DMAPA with different ECH-to-DMAPA molar ratio(reaction temperature=55℃,reaction time=2 h)

图9 ESI-MS图谱对应的可能结构Fig.9 Possible chemical structures associcated to the peaks found in ESI-MS spectra of CPILs
ESI-MS 测试中观察到了质荷比(m/z)为261.33、419.50 和577.58 的峰,分别可能对应[C13H33N4O]+、[C21H51N6O2]+和[C29H69N8O3]+,其可能的结构如图9 所示。FT-IR 和ESI-MS 的表征结果均与图7 中提出的反应机理吻合。
2.4 CO2渗透性能测试
按照探索的反应条件和方法,合成了低交联度的ECH0.5-[DMAPAH][MOAc],并以此为基础制备支撑液膜,探究交联对其CO2分离性能的影响。如图10 所示,随着CO2分压的增加,CO2渗透系数呈现下降的趋势,这是典型的促进传递机理[27,29]。在40℃和0.025 bar分压的含水条件下,CO2渗透系数为594 barrers,CO2/N2选 择 性 为567(N2渗 透 系 数 为1.05 barrers),该结果远高于文献中报道的RTILs 以及绝大多数TSILs 的分离性能(CO2/N2选择性一般不超过200[31]),表明该类交联质子型离子液体适合低浓度CO2的高选择性分离。与未交联的[DMAPAH][MOAc]分离性能(40℃、0.025 bar 和15%(质量)含水条件下CO2渗透系数为2100 barrers,CO2/N2选择性为100)相比,交联后CO2的渗透系数明显下降,这可能是由于ECH与[DMAPAH][MOAc]交联消耗了部分游离胺基,导致对CO2的促进传递能力的削弱[27];同时,交联后CO2/N2的选择性大幅提高,这可能是由交联生成的氯离子和铵离子产生的盐析效应导致的。在后续的工作中将继续优化该类交联质子型离子液体的CO2分离性能。

图10 40°C含水条件下ECH0.5-[DMAPAH][MOAc][含水20%(质量)]中CO2的渗透系数及CO2/N2选择性随跨膜压差的变化关系Fig.10 Plots of permeability of CO2 and CO2/N2 selectivity in ECH0.5-[DMAPAH][MOAc]of 20%(mass)water as a function of transmembrane pressure difference under humidified condition at 40℃
3 结 论
针对离子液体凝胶化过程中使用的交联剂依然存在价格高昂、制备困难等缺点,选用了一种便宜易得的交联剂——环氧氯丙烷,探究其与一种具有高CO2分离性能的二胺有机酸盐质子型离子液体([DMAPAH][MOAc])交联的可能性。动力学测试结果表明,先交联再与酸中和有利于提高环氧氯丙烷的转化率;其次,当ECH 与DMAPA 摩尔比为1∶1时,在55℃条件下反应2 h 后氯基团几乎100%转化,环氧基团的转化率也高达70%。FT-IR 和ESIMS表征也证明了反应机理。此外,通过在线监控反应体系黏度变化的方法发现,在55℃和乙二醇为溶剂的条件下,体系在第70 min 左右出现凝胶点,证明这类环氧氯丙烷-二胺交联产物具有很好的凝胶转变性能。气体渗透初步测试结果也表明该类交联产物具有制备高CO2选择性分离膜的潜力。该工作为环氧氯丙烷交联的质子型离子液体膜材料的制备奠定了初步的理论基础。
