高稳定甲烷干重整Ni基催化剂的制备及其催化性能研究
2020-11-16黄艳丽李晓东
黄艳丽,李晓东,黄 伟*
(1.太原理工大学 煤科学与技术教育部和山西省重点实验室,山西 太原 030024;2.晋中学院 化学化工学院,山西 晋中 030619)
由于页岩气回收技术的进步,甲烷二氧化碳重整(CH4+CO2→2H2+2CO),也称为甲烷干重整(DRM),引起了广泛的关注。DRM过程具有环保和经济双重意义,因为在该反应过程中,两种主要的温室气体(CH4和CO2)被转化为具有工业价值的合成气(CO和H2),且由于逆水煤气变换反应(RWGS:CO2+H2→CO+H2O),n(H2)/n(C)始终低于1[1-3]。 用于该反应的催化剂有Rh、Pt、Ir、Pd、Ru、Co和Ni。 与Ni基催化剂相比,贵金属催化剂表现出了更好的抗积炭性能,然而贵金属催化剂的高成本限制了其大规模使用,因此开发高稳定的Ni基催化剂成为该课题的研究重点[4]。
将Ni-Mg-Al类水滑石通过煅烧和还原预处理制得的Mg(Ni,Al)O复合氧化物催化剂具有大的表面积、适度的碱性、良好的热稳定性以及较小的Ni晶粒尺寸[5-7]。在课题组前期的研究中,首先通过焙烧Ni-Mg-Al类水滑石前驱体,制备了高稳定的Ni/MgAl(O)催化剂,该催化剂可使CH4和CO2的转化率维持在95%以上2000 h,且反应后催化剂表面积炭很少,仅有0.0351 mg/(g·h)[8]。 此外,许多助剂(如:Mo、Ce、Zr、La)被用于提高以NiMgAl类水滑石为前驱体制备的Ni基催化剂的稳定性[9-14]。
类水滑石的合成方法有共沉淀法、水热法、浸渍法和熔胶凝胶法[15]。水热法可以避免杂质的引入,因为反应是在密闭的水热釜中进行的,该方法还可以避免引入对催化性能不利的碱金属离子(Na+、K+),因此由该方法制备的催化剂更容易洗涤;用于水热法的沉淀剂有尿素、氨水和六次甲基四胺(HMT),而HMT在制备均匀细小的结晶性粉末方面具有明显的优势,因此被广泛用作控制颗粒大小和形貌的沉淀剂[16,17]。本文拟采用简单的均匀沉淀法,通过调控n(HMT)/n(Ni+Mg+Al)比值,制备Ce、Zr和Mo改性的NiMgAl类水滑石前驱体,并将焙烧后的Ni/MoCeZr-MgAl(O)复合氧化物催化剂用于CH4-CO2重整实验中,考察不同n(HMT)/n(Ni+Mg+Al)对催化剂性能的影响。
1 实验部分
1.1 催化剂的制备
将一定量的Ni(NO3)2·6H2O、Mg(NO3)2·6H2O、Al(NO3)3·9H2O、(NH4)6Mo7O24·4H2O、Ce(NO3)2·6H2O和ZrO(NO3)2·xH2O溶解于150 mL去离子水中,其中n(Ni2++Mg2+)/n(Al3+)=3。称取一定量的HMT固体(使得n(HMT)/n(Ni+Mg+Al)为1、2、2.5或3.5)溶解于200 mL去离子水中。将两种溶液转移至一个烧杯中,并持续搅拌2 h,转至不锈钢高压水热釜中110℃下晶化1 d,然后冷却、过滤、洗涤、干燥(80 ℃,12 h),程序升温至800 ℃(5℃/min),恒温焙烧6 h。制得的催化剂中NiO、(ZrO2+CeO2)和MoO3的质量分数分别为10%、1%和0.6%;根据制备时不同的n(HMT)/n(Ni+Mg+Al),将催化剂分别命名为S1、S2、S2.5、S3.5。
1.2 催化剂的表征
1.2.1 X射线衍射(XRD)
采用DX-2700型X射线衍射仪 (Cu Kα射线源,管电压40 kV), 以8(°)/min的扫描速率和0.01°的步长,从5°~85°对催化剂的晶相结构进行了测试。
1.2.2 N2物理吸-脱附
样品的比表面积和孔径分布在美国Micromeritics公司的ASAP 2020系列吸附仪上测定,所有样品造粒(40~60目),称取0.1 g催化剂,200 ℃脱气处理4 h,然后进行N2吸-脱附实验,并计算催化剂的平均孔径(BJH模型)和比表面积(BET方程)。
1.2.3 程序升温还原(H2-TPR)
在天津先权公司TP-5000化学吸附仪上进行程序升温还原实验。50 mg样品装入石英管中间,在150℃氦气气氛下,恒温吹扫0.5 h,然后降温至50℃后,切换为5%H2-N2混合气(流速20 mL/min),然后以10℃/min的升温速率升至900℃,热导检测器(TCD)检测H2信号,以氧化银(Ag2O)作为标准,并计算氢气的消耗量以及催化剂的可还原程度。
1.2.4 CO化学吸附
在ChemiSorb2720化学吸附仪(美国Micromeritics公司)上进行CO脉冲吸附实验,称取130 mg样品(40~60目)装入U型石英管中,10%H2-Ar气氛下900 ℃处理2 h,待温度降至50℃后,10%CO-He脉冲进样,每次所进的CO体积为0.2 mL,TCD检测经样品吸附后残余的CO量,当色谱中的峰面积恒定时,即可判断催化剂吸附达到饱和,进而算出样品吸附的CO量。假定n(CO)/n(Ni(s))=1,根据催化剂的CO吸附量以及Ni原子的横截面积(6.5×10-20m2),得到样品中的Ni的分散度(%)。
1.2.5 程序升温脱附(CO2-TPD-MS)
CO2-TPD-MS实验在天津先权公司生产的TP-5080型程序升温吸附仪上进行。称量100 mg还原后催化剂装入石英管中,He气氛下以10℃/min的升温速率升至280℃恒温吹扫30 min;降温至50℃后,通入CO2气体连续吸附30 min;然后将气体切换为He气并吹扫30 min。He气氛下以10℃/min的升温速率升至900 ℃进行脱附,质谱(HIDEN,QIC-20)检测CO2的信号。
1.2.6 程序升温氧化(O2-TPO-MS)
为了测定催化剂表面积炭的种类以及积炭量,在TP-5080程序升温吸附仪(天津先权公司)上进行程序升温氧化实验。先称量反应后的催化剂100 mg,然后装入石英管中间部位(两端用石英棉固定),He气氛以10℃/min的升温速率升至150℃吹扫30 min;然后降温至50℃后,通入5%O2-Ar的混合气,并保持30 min然后,以10℃/min的升温速率从50℃升至900℃进行程序升温氧化反应,质谱仪(HIDEN,QIC-20型)检测CO2的信号,以CO2信号与温度作图,通过积分定量计算催化剂表面的积炭量(活性C为标准)。
1.3 催化剂的性能测试
在常压固定床反应器上测定了催化剂的活性和稳定性。所有催化剂和石英砂的尺寸为40~60目,称量300 mg催化剂与1.5 g石英砂并混合均匀,转移至石英管反应器内,催化剂两端用石英棉固定,然后将反应器置于加热炉中,使催化剂位于反应炉的恒温区;催化剂的还原采用程序升温还原,H2-N2(N2流速为20 mL/min、H2流速为20 mL/min)气氛下,以5℃/min的升温速率升温至450℃,然后以1℃/min的升温速率从450℃升温至900℃,在900℃恒温还原120 min,然后通入n(CH4)/n(CO2)=1的原料气,流速为150 mL/min,GHSV=60000 mL/(g·h)。 使用上海海欣公司的GC-950气相色谱仪检测并分离尾气中的CH4、H2、CO和CO2,检测器为TCD,载气是Ar气,流速为35 mL/min,色谱柱为填充柱(TDX-01),采用外标法分别计算产物(CO、H2)的选择性和原料气(CH4、CO2)的转化率。柱温130℃,TCD温度100℃,汽化室温度150℃,热导桥流为40 A,进样1 mL。
2 结果与讨论
2.1 催化剂的催化性能
不同n(HMT)/n(Ni+Mg+Al)制备的催化剂在900℃的甲烷二氧化碳重整反应结果见图1。从图1(a)可以清楚地看到催化剂S2、S2.5和S3.5的初始活性(CH4转化率)都与该温度下热力学平衡转化率接近(97%)。S1催化剂的初始活性较低,CH4转化率为92.7%,且只经过24 h重整反应后降至89.3%;S2、S2.5和S3.5分别经过600 h、36 h和150 h反应后,CH4的转化率变至92.3%、91.5%和90.4%。值得注意的是,S2催化剂的活性呈现出一个波动的趋势,即CH4转化率,CO2转化率,H2与CO物质的量比呈现出一个波动趋势,且该趋势维持了长达600 h。对比图1(a)和图1(b)可以发现,对于所有催化剂,CO2的转化率略高于CH4的转化率,这是由于RWGS反应的发生,该反应消耗了H2并生成了CO;图1(c)中H2与CO物质的量比始终低于化学计量比(1:1),也证明了这一点。

图1 催化剂的评价结果:(a)CH4转化率,(b)CO2转化率和(c)H2与CO物质的量比Fig.1 Catalyst evaluation results:(a)the conversion of CH4,(b)the conversion of CO2and(c)the molar ratio of H2/CO
2.2 催化剂的表征
2.2.1 X射线衍射和N2物理吸脱附结果分析
图2(a)为未焙烧催化剂的XRD谱图,从图中可以看出只有S1催化剂的XRD谱图为物相单一的类水滑石特征衍射峰(003)、(006)、(009)、(015)、(018)、(110)和(113),而其他催化剂的前驱体都出现了Mg(Ni)CO3的特征衍射峰。图2(b)是焙烧后催化剂的XRD图谱,焙烧后催化剂的XRD谱图中都出现了Mg(Ni,Al)O固溶体和CeO2(2θ=28.9°和47.4°)的特征衍射峰。 另一个现象是在所有样品的XRD谱图中,都没有发现ZrO2和MoO3物种的特征衍射峰,这可能与其较低的负载量或弱的结晶度有关,超出了XRD的检测限。
图3(a)和(b)分别是催化剂还原后和反应后的X射线衍射图。对比图3(a)和(b)可以发现还原后和反应后的催化剂都呈现了相似的物相,其中2θ=36.8°、43.5°、62.7°和79.1°的峰归属于Mg(Ni,Al)O方镁石的特征衍射峰,2θ=19.0°、31.5°、44.9°、59.6°和65.5°处的衍射峰为Ni(Mg)Al2O4尖晶石的特征衍射峰,2θ=28.9°和47.4°为CeO2的特征衍射峰 ,2θ=44.5°、51.8°和76.3°是Ni的特征衍射峰。值得注意的是,反应后S1、S2和S3.5催化剂的XRD图谱中, 在2θ=26.8°处的特征衍射峰归属为石墨碳的衍射峰,O2-TPO-MS表征结果也证明了这一点。对比图3(a)和(b),Ni的特征衍射峰(2θ=44.5°、51.8°和76.3°)的衍射强度在经过DRM反应后都呈现出不同程度的增强趋势,这反映了催化剂的Ni晶粒在反应过程中发生了一定程度的团聚。通过Scherrer公式,计算了Ni(200,2θ=51.8°)的晶体尺寸,见表1。

图2 (a)前驱体、(b)焙烧后催化剂的XRD图谱Fig.2 XRD profiles of(a)the precursors and(b)the calcined catalysts
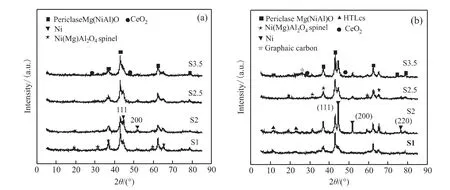
图3 (a)还原后催化剂、(b)反应后催化剂的XRD图谱Fig.3 XRD profiles of(a)the reduced and(b)the spent catalysts

表1 焙烧和反应后催化剂的比表面积、耗氢量、Ni2+还原程度、Ni的分散度以及Ni的晶粒尺寸Table 1 Textural properties of the calcined and spent catalysts surface area,H2consumption,Ni2+reduction degree,Ni dispersion and Ni particle size
可以看出还原后S1、S2、S2.5和S3.5催化剂的Ni晶粒尺寸分别为 7.4 nm、7.5 nm、8.0 nm和7.8 nm,而反应后的催化剂的晶粒尺寸增大至11.0 nm、20.8 nm、10.9 nm和12.7 nm,都呈现出不同程度的增大趋势。
由焙烧后催化剂的N2物理吸-脱附实验结果可知,相关曲线都呈现出IV型等温线,表明所有样品都是典型的介孔结构,同时均显示出H3型回滞环,表明孔的类型主要是由片状粒子堆积形成的狭缝孔。表1总结了焙烧后催化剂详细的织构性质,所有催化剂的比表面积都在51~160 m2/g范围内,这是类水滑石来源材料的典型特征,且S2催化剂的比表面积最大。
2.2.2 程序升温还原和CO化学吸附结果分析
图4为800°C煅烧后催化剂的H2-TPR谱图。S1、S2、S2.5和S3.5催化剂在700~880 °C的高温区域均观察到一个还原峰,结合焙烧后催化剂的XRD结果(图2b),该峰可归属于Mg(Ni,Al)O方镁石中Ni2+的还原,与纯的NiO(370~450 °C)还原温度相比,高温还原峰(700~880°C)表明样品中NiO物种与载体之间存在较强的相互作用力。通过积分计算了耗H2量和Ni2+的还原程度 (表1),S1、S2、S2.5和S3.5的还原度均小于100%,这是由于Ni2+物种是以Mg(Ni,Al)O固溶体的形式存在,因此部分NiO物种难以被还原[10]。值得注意的是,S2催化剂的还原温度最低(783°C)、耗H2量最大、可还原物种最多,这反应了其活性组分分散度最好,这与CO化学吸附表征结果一致。从表1可以看出,S2催化剂Ni的分散度最大(15.7%),结合催化性能测试结果,这可能是其具有最佳催化性能的原因。

图4 煅烧后催化剂的H2-TPR谱图Fig.4 H2-TPR profiles of the calcined catalysts
2.2.3程序升温脱附结果分析
图5为900°C还原后Ni/MoCeZr-MgAl(O)催化剂的CO2-TPD-MS谱图。所有催化剂均在101℃、199℃和300~550℃出现三个峰,说明催化剂表面存在三种碱性位,不同比例的HMT使用对催化剂碱中心的类型没有显著的影响。催化剂的碱性位越强,CO2的解吸温度越高。低强度(101℃)和中等强度(199℃)的碱性位点分别对应于表面OH和Lewis酸碱对的CO2解吸,高强度(300~550℃)的碱性位点与低配位点表面O2-与CO2的吸附有关[18]。 此外,碱位点数(见表2)随着n(HMT)/n(Ni+Mg+Al)的增加整体上呈现出增大的趋势,这可能是由于HMT的增加使催化剂中Mg元素的含量增加,MgO能促进CO2分子在催化剂上的吸附,从而增加催化剂表面碱性中心的数量[19]。

图5 还原后催化剂的CO2-TPD-MS图谱Fig.5 CO2-TPD-MS profiles of the reduced catalysts
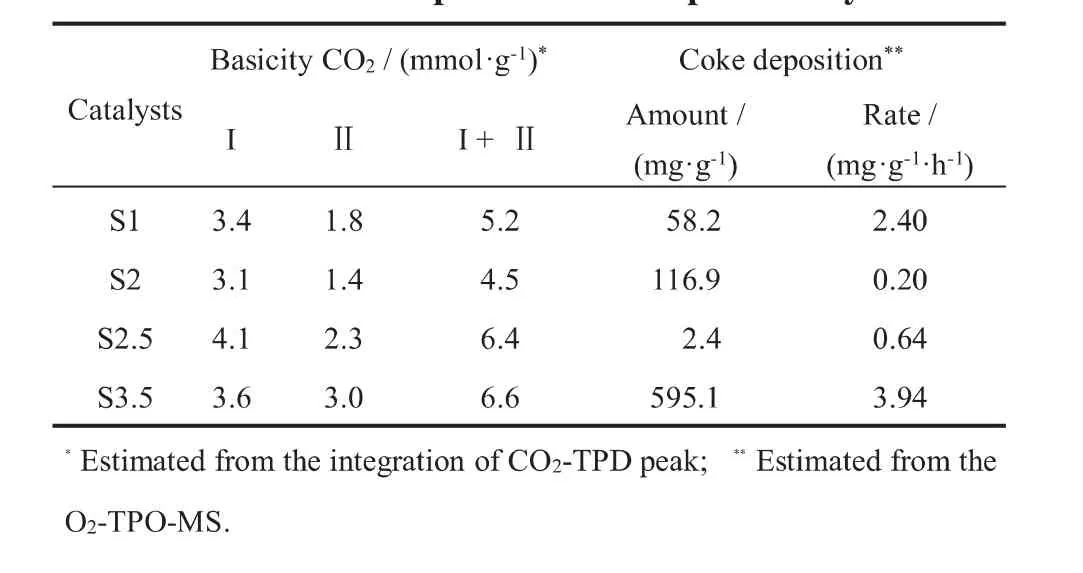
表2 还原后催化剂的碱性位点数以及反应后催化剂的积炭Table 2 The basic properties of the reduced catalysts and coke deposition of the spent catalysts
2.2.4 程序升温氧化结果分析
采用程序升温氧化(O2-TPO-MS)实验研究了催化剂表面积炭种类和积炭量。催化剂表面的积炭按照其氧化温度可分为如下三类:Cα(<250 °C),Cβ(300~500 °C),Cγ(>600 °C), 相对于Cα和Cβ,Cγ更难被氧化,是造成催化剂失活的主要碳物种[20]。如图6所示,S1、S2和S3.5催化剂的O2-TPO曲线非常相似,主要积炭的种类是石墨碳(Cγ),而S2.5催化剂的主要积炭种类为Cβ。以活性炭为基准,通过积分定量计算了催化剂表面的积炭总量(见表2)。在900°C反应600 h后,S2催化剂在催化剂表面只有少量的积炭生成(0.20 mg/(g·h)),这说明该条件下制备的催化剂具有较好的抗积炭性能。

图6 反应后催化剂的O2-TPO-MS图谱Fig.6 O2-TPO-MS profiles of the spent catalysts
3 结论
通过简单的一步水热法成功制备了Ce、Zr和Mo改性的NiMgAl类水滑石前驱体,经过800℃焙烧和900℃还原处理后,获得一系列Ni/MoCeZr-MgAl(O)复合氧化物催化剂,并将该催化剂用于DRM反应中,结果表明催化剂的催化性能与n(HMT)/n(Ni+Mg+Al)有关,其中,S2催化剂表现出最佳的稳定性,在900℃,空速为60000 mL/(g·h),n(CH4)/n(CO2)=1时,反应600 h后催化剂的甲烷转化率仍能保持在93%以上,且积炭速率仅有0.20 mg/(g·h)。由表征结果可知较为单一的类水滑石晶相,高的金属分散度,大的表面积以及适宜的碱性位点数是其高活性和稳定性的原因。
