1个复合杂合突变导致的凝血因子Ⅺ缺陷症家系表型及基因型分析
2020-11-06毕晓洁黄道超金先富姜俊宇苏正仙陈超超
毕晓洁, 黄道超, 金先富, 姜俊宇, 苏正仙, 陈超超, 沈 波
(1.温州医科大学附属台州医院检验科,浙江 临海 317000;2.温州医科大学附属台州医院急诊科,浙江 临海 317000)
遗传性凝血因子(coagulation factor,F)Ⅺ缺陷症为罕见的常染色体隐性遗传病,于1953年首次被ROSENTHAL等[1]报道,目前,突变类型已超过200多种(http://www.hgmd.cf.ac.uk/ac/gene.php?gene=F11),国外统计的发生率为1/500 000~1/1 000 000,不同发病率存在种族差异[2]。我国对于遗传性FⅪ缺陷症的报道较少,具体的致病机制也不是很明确。本研究对1例遗传性FⅪ缺陷症进行表型和基因型研究,探究其分子致病机制。
1 材料和方法
1.1 研究对象
1.1.1 家系资料 该家系共8人,包括先证者及父亲、母亲、姐姐、姐夫、外甥女、儿子、妻子。家系谱见图1。先证者,男,36岁,因反复鼻衄入院。实验室常规凝血功能检查示:活化部分凝血活酶时间(activated partial thromboplastin time,APPT)显著延长,FⅪ活性(FⅪ:C)极低,D-二聚体(D-dimer,DD)轻度升高,其余指标均在参考区间内。先证者肝、肾功能正常,未使用抗凝药物。先证者父母非近亲结婚,其他家系成员均无自发出血或血栓形成病史。
1.1.2 正常对照组 以140名体检健康者作为正常对照组,其中男72名、女68名,年龄40(21~62)岁。无肝、肾疾病,无血栓或出血史。

图1 遗传性FⅪ缺陷症家系图
1.2 仪器与试剂
1.2.1 仪器 STA-R全自动血凝仪(法国STAGO公司)、AU5830全自动生化分析仪(美国雅培公司)、ABI Thermal cycler 2720 PCR扩增仪(美国ABI公司)、核酸蛋白分析仪(美国Beckman Coulter公司)、电泳仪(美国Bio-Rad公司)、凝胶成像系统(上海天能公司)。
1.2.2 试剂 凝血项目检测试剂盒购自法国STAGO公司。DNA提取试剂盒、抗原检测试剂盒及聚合酶链反应(polymerase chain reaction,PCR)试剂盒均购自上海西塘生物科技技术公司。
1.2.3 引物 引物序列参考文献[3],由华大基因生物公司合成。
1.3 方法
1.3.1 标本采集和处理 采集所有受试者外周血,用0.109 mol/L枸橼酸钠1∶9抗凝。上层血浆用于凝血项目检测;下层血细胞用于提取DNA,提取完成后置于-20 ℃冰箱保存待用。
1.3.2 凝血项目检测 在全自动血凝分析仪上采用一期凝固法检测凝血酶原时间(prothrombin time,PT)、APTT、纤维蛋白原(fibrinogen,Fib)、凝血酶时间(thrombin time,TT)、FⅡ活性(FⅡ∶C)、FⅤ活性(FⅤ∶C)、FⅦ活性(FⅦ∶C)、FⅧ活性(FⅧ∶C)、FⅨ活性(FⅨ∶C)、FⅩ活性(FⅩ∶C)、FⅪ活性(FⅪ∶C)及FⅫ活性(FⅫ∶C);免疫比浊法检测纤维蛋白(原)降解产物[fibrin(fibrinogen)degradation product,FDP]、DD;采用酶联免疫吸附试验检测FⅪ抗原(FⅪ∶Ag)水平。
1.3.3 抽提外周血基因组DNA 使用DNA提取试剂盒提取所有成员以及正常对照组外周血基因组DNA。
1.3.4 PCR扩增及电泳 PCR反应总体积为50 μL,其中DNA模板3 μL、PCR MaserMix 25 μL,双蒸水18 μL,上、下游引物各2 μL。反应条件:95 ℃预变性5 min;95 ℃变性30 s,58~60 ℃退火30 s,72 ℃延伸1 min,30个循环;72 ℃延伸10 min。
1.3.5 DNA测序及分析 PCR产物进行直接测序,测序结果采用Chromas软件与GenBank中的基因序列进行比对,发现突变后进行反向测序予以验证。
1.3.6 生物学信息软件分析 采用Mutation Taster(http://www.mutationtaster.org)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)和SIFT(http://sift.jcvi.org)3个在线生物信息软件,通过评分预测突变位点对蛋白质功能的影响。
2 结果
2.1 凝血指标检测结果
先证者以及家系成员凝血功能检测结果见表1。
2.2 基因分析结果
先证者F1 1基因1 1号外显子存在c D N A.1 6 4 0 G>A,1 4号外显子存在cDNA.2183G>A,导致Ala430Thr和Val611Met复合杂合突变。见表2、图2和图3。
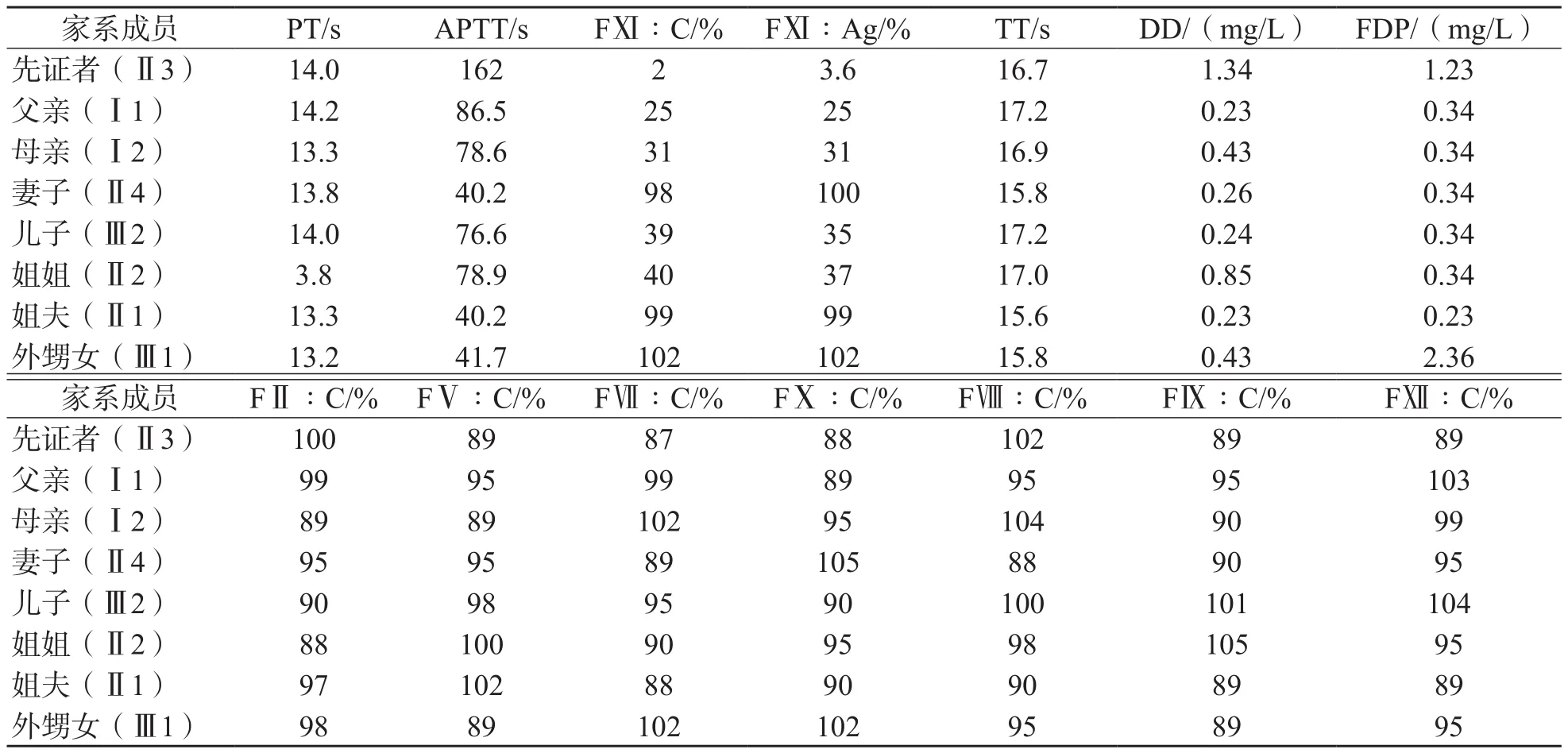
表1 先证者及家系成员凝血指标检测结果

表2 先证者及家系成员基因突变检测结果
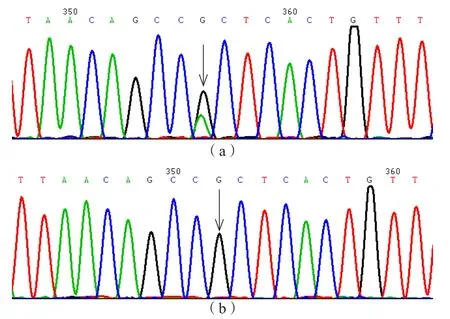
图2 先证者及父亲F11基因11号外显子Ala430Thr突变及野生型

图3 先证者、母亲、儿子、姐姐F11基因14号外显子Val611Met突变及野生型
2.3 生物学信息软件分析结果
PolyPhen-2评分为1.00分,提示此突变为有害突变;Mutation Taster评分结果为disease causing,提示此突变可引起疾病;SIFT评分为0.10分,提示此突变可引起蛋白质功能改变。
3 讨论
FⅪ是一种丝氨酸蛋白酶原,相对分子质量为16 000,在血浆中其主要与高分子量激肽原以非共价形式结合形成复合物[4]。编码FⅪ的基因F11位于4号染色体长臂(4q35),共含有15个外显子和14个内含子,全长23 kb[5],l号外显子编码的是5'端非转录区,2号外显子编码前导肽链,3-15号外显子共编码607个氨基酸分子,形成的FⅪ包括含有4个Apple结构域的重链以及1个胰蛋白酶样丝氨酸蛋白酶催化区的轻链。成熟的FⅪ分子含有2个相同单体,由2个残基Cys321形成二硫键组成[6]。
根据经典凝血瀑布学说原理,FⅪ在内源性凝血途径中发挥重要作用。FⅪ在凝血过程中被活化的FⅫ、凝血酶或自身裂解所激活,生成活化的FⅪ[7],再通过一系列的凝血因子逐级活化过程,最终激活纤维蛋白原,形成纤维蛋白网,使血小板形成的团块更加牢固[8]。FⅪ缺陷临床表现多样,突变基因杂合子的血浆FⅪ水平为正常人的30%~70%,而纯合子的血浆FⅪ水平仅为正常人的0~20%[9]。该病患者无性别差异,与血友病A和B相比,出血症状相对较轻,且一般无自发性出血,出血常与创伤和手术相关[10],因此疾病的诊断和治疗都存在多变性。原因可能是FⅪ参与凝血过程的放大阶段而不是影响凝血的起始阶段,并且活化的血小板颗粒含有FⅪ,总活性得到一定的补偿,因此FⅪ缺陷症患者多数是在手术、创伤后才会表现出止凝血障碍[11]。而出血的严重程度常与累及的组织部位有关,原因是FⅪ还可引起凝血酶激活纤溶抑制物缺乏,因此在纤溶活性高的组织更容易出血,常见于黏膜[12]。
本研究中的先证者APTT延长至162 s,APTT纠正试验可以纠正,排除因抑制物引起的获得性凝血因子异常;FⅪ∶C为2%,FⅪ∶Ag为3.6%,活性与抗原成等比例下降,呈交叉反应物质阴性特点。先证者父亲、母亲、姐姐及儿子均存在不同程度的FⅪ∶C和FⅪ∶Ag等降低。基因测序结果显示,先证者F11基因的11号外显子存在cDNA.1640G>A,14号外显子存在cDNA.2183G>A,导致Ala430Thr和Val611Met复合杂合突变,分别来自于父亲和母亲,可以诊断为遗传性FⅪ缺陷症。
11~15号外显子编码的是多肽链的羧基端[14],是1个胰蛋白酶样丝氨酸蛋白酶结构域,与FⅪ活化有关,发生在该区域的突变会导致空间构象产生变化,最终使多肽链的折叠发生异常,蛋白质的稳定性变差,极易被降解,FⅪ活化发生障碍。Ala430位于α螺旋结构内部,丙氨酸被苏氨酸替代后,与其周围的Phe433和Tyr436空间位阻发生改变,α螺旋结构稳定性变差[13]。该研究中患者FⅪ∶C为48.6%,FⅪ∶Ag为50%,无任何出血症状,与本研究先证者父亲的表型类似,14号外显子存在cDNA.2183G>A,该突变是目前国际上发现的新的突变位点。611号缬氨酸突变为甲硫氨酸,分子链变短,与周围氨基酸的作用力发生改变,蛋白质稳定性变差。
综上所述,Ala430Thr和Val611Met复合杂合突变是导致本例患者FⅪ∶C明显下降的主要原因。但具体作用机制尚待进一步研究加以证实。
