CLDN16基因突变致家族性低镁血症高钙尿症伴肾钙质沉着症基因型与表型特点分析
2020-11-06热衣兰木包尔汉罗燕飞孙光辉迪丽胡麻居来提米热古丽买买提
热衣兰木·包尔汉, 李 燕, 罗燕飞, 孙光辉, 迪丽胡麻·居来提, 米热古丽·买买提
(新疆医科大学第一附属医院,新疆 乌鲁木齐 830054)
家族性低镁血症高钙尿症伴肾钙质沉着症(familial hypomagnesemia with hypercalciuria and nephrocalcinosis,FHHNC)是一种罕见的常染色体隐性遗传病,由编码肾小球髓袢远端紧密连接蛋白claudin-16的CLDN16基因或蛋白claudin-19的CLDN19基因突变所致。FHHNC以肾脏排泄镁、钙过多导致的低镁血症、高钙尿症、双肾钙质沉着症及进行性慢性肾功能衰竭为特征[1]。本研究回顾性分析1例以严重佝偻病为主要表现的FHHNC患儿,并结合相关文献,总结其基因型与表型特征。
1 材料和方法
1.1 研究对象
患儿,女,11岁,维吾尔族,2019年1月因无明显诱因出现抽搐就诊于当地医院,抽搐形式为四肢强直痉挛,呈角弓反张样,无双眼上翻,无意识丧失,无大小便失禁,持续数十秒至1 min后缓解,无乏力、嗜睡。完善腹部彩超及泌尿系统电子计算机断层扫描(computed tomography,CT),提示“双肾结石”,行体外冲击波碎石术1次,效果欠佳,患儿结石未排除,但无泌尿系统相关症状,因此未作进一步治疗。后因患儿仍间断抽搐,一直不能行走,被动坐位。为明确病因,于2019年6月27日至新疆医科大学第一附属医院进一步诊治。本研究经患儿家长知情同意并签署书面同意书。
1.2 方法
收集患儿的临床资料及相关检查结果,包括X线、B超、CT、磁共振成像(magnetic resonance imaging,MRI)、脑电图及实验室检测项目。采集患儿及其父母外周血各2 mL,进行全外显子组基因测序分析。将检测范围内相关基因的编码区使用基于目标区域捕获的方法进行扩增,并采用Illumina二代测序平台(美国Illumina公司)测序,解读软件为bpvastg-TIES基因检测智能系统,根据致病XHGMD数据库专业版(2017)和CliVariation数据库评估基因突变的致病性。
2 结果
2.1 体格检查
患儿生命体征平稳,发育落后。身高为90 cm[低于同年龄、同性别、同地区儿童平均身高8.58个标准差(-8.58s)],体质量为16 kg。面容:鼻梁塌平,眼距宽,颈短。心肺查体未见异常,腹部膨隆,腹围62 cm,无腹肌紧张,无压痛及反跳痛。鸡胸,四肢弯曲畸形,肘关节及膝关节膨大,被动盘腿坐位,四肢末梢循环良好,神经系统查体无异常。见图1。
2.2 家系遗传系谱图
患儿为第4胎第4产,足月顺产,出生体质量为3 900 g,出生3个月时抬头,出生10个月时可坐,至今不会行走。患儿喂养史无异常,其父母为近亲结婚,系表兄妹,家系遗传系谱图见图2。患儿姐姐(Ⅱ3)平日多次出现无热抽搐,抽搐形式与本例患儿类似,于6岁时因突发抽搐,四肢强直痉挛,呈角弓反张样,口唇发绀,家长诉患儿出现喉部喘鸣后呼吸停止,死亡。家族其他亲属均无类似泌尿系统、骨骼系统及神经系统症状。

图1 本例患儿体格检查特征

图2 本例患儿家系遗传系谱图
2.3 实验室、影像学及其他检查结果
2.3.1 实验室检测结果 血钙1.21 mmol/L(降低),血镁0.4 mmol/L(降低),尿钙4.11 mmol/L(升高),25-羟基维生素D 23.62 nmol/L(降低),血清骨钙素191.4 ng/mL,甲状旁腺激素63.3 pmol/L。肌酐、尿酸及尿素氮均正常。尿常规:pH值6.0,白细胞计数69个/μL,细菌计数121个/μL。多次尿培养:弗氏柠檬酸杆菌,接种10 μL培养,菌量>105CFU/mL。血常规、甲状腺功能、血气分析及肾小管功能相关项目检测均未见明显异常。
2.3.2 影像学及其他检查 眼科检查未见明显异常。胸部正位片:胸廓骨骼变形,密度增高,考虑代谢性改变;心影增大。手部正位片:左腕见8腕骨,左手及腕部诸骺线未闭合,骨质疏松,考虑遗传代谢病。本例患儿四肢X线片见图3。心脏及甲状旁腺超声未见异常。泌尿系统超声检查结果:双肾钙质沉着,多发结石,大小约为1.0 cm,较大者位于中盏,双侧输尿管未见扩张,膀胱未见明显异常,见图4。泌尿系统及骨骼三维重建:双肾集合系统多发高密度填充,考虑结石;所及肋骨、胸椎、腰椎、骶骨及双侧髋关节骨质密度增高,变性,见图5。头颅MRI检查结果:双侧尾状核、苍白球及壳核对称性异常信号;扫及颅骨及颈胸椎诸椎体及附件骨质弥漫性减低,颅骨板障增厚,斜坡及寰椎形态异常,考虑代谢性疾病所致可能性大。脑电图:正常儿童脑电图。
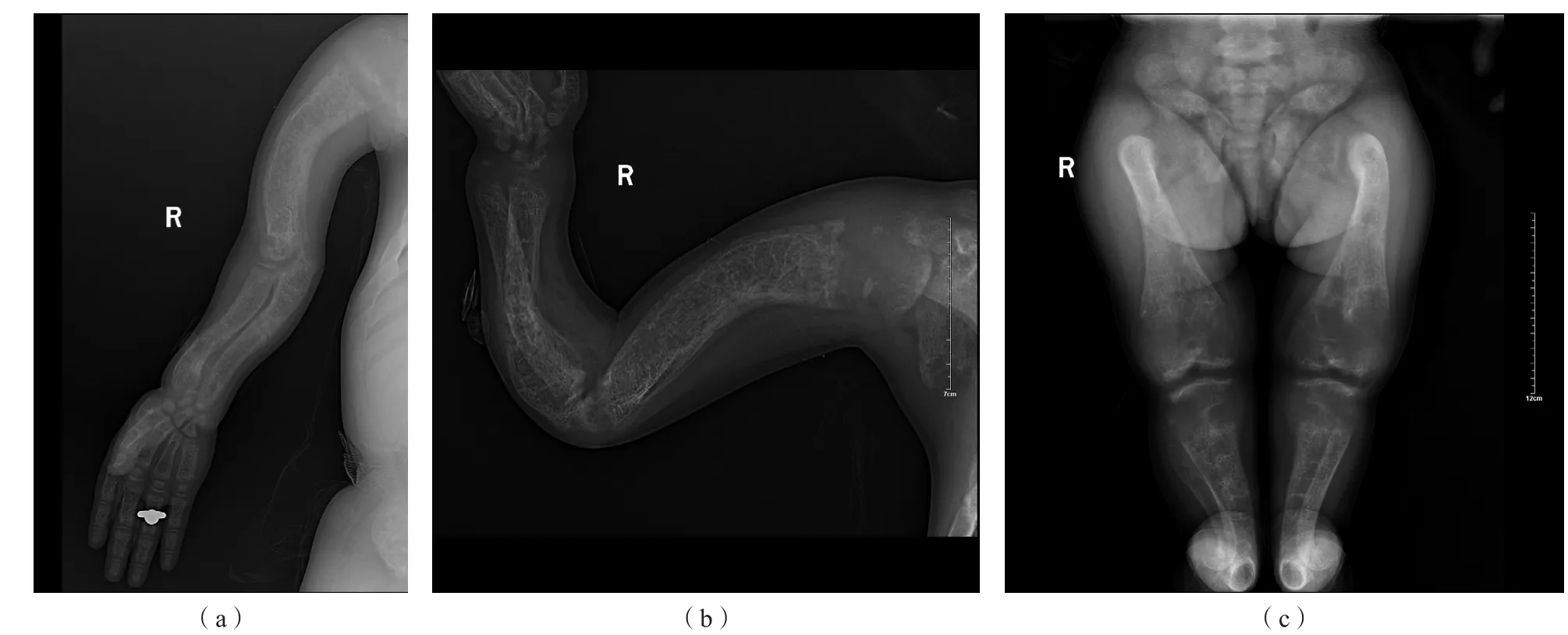
图3 本例患儿四肢X线检查结果

图4 本例患儿泌尿系统B超图像

图5 本例患儿泌尿系统及骨骼三维重建
2.4 基因检测
结合患儿病史、查体结果及辅助检查结果,患儿有低血镁、高尿钙、肾钙质沉着,同时合并抽搐及骨骼系统畸形,涉及多个系统异常,考虑遗传性代谢病,FHHNC可能性大,建议行基因检测。基因测序结果显示,患儿CLDN16基因(NM_006580.3)存在“C.647G>A,p.Arg216His”[Chr3(GRCh37):g.190126157G>A]纯合致病突变,患者父母均携带该位点变异(杂合),见图6。

图6 本例患儿及其父母全外显子基因测序图
2.5 治疗及预后
患儿确诊为FHHNC后,嘱其低盐饮食,多饮水,住院期间补镁、补钙,出院后继续给予补钙、骨营养支持治疗。因患儿双肾结石的手术难度大,术后易复发,且患儿肾功能正常,结石也未引起梗阻,因此暂不考虑手术治疗。针对患儿泌尿道感染情况,根据药物敏感性试验结果给予抗菌治疗,每月查尿常规、尿培养及血、尿电解质,注意清洁。随访6个月,患儿低血镁、高尿钙等症状明显好转,双肾钙质沉着无加重,肾功能正常,25-羟基维生素D水平在参考区间内。手足抽搐和肌无力症状完全缓解,出院后未出现抽搐,已可扶走。
3 讨论
FHHNC是一种罕见的常染色体隐性遗传病,由MICHELIS等[2]在1972年首次报道,其致病基因为CLDN16和CLDN19,分别编码肾小球髓袢远端紧密连接蛋白claudin-16和claudin-19。claudin-16和claudin-19是包含4个跨膜结构域和2个细胞外段的膜蛋白家族成员之一,二者相互作用构成异二聚体,组成选择性阳离子通道,镁和钙在跨上皮电位驱动下通过细胞旁途径被重吸收[3]。当CLDN16和CLDN19发生突变时,肾脏对镁、钙的重吸收减少,从而出现低镁血症、高钙尿症、双侧肾钙质沉着和进行性肾功能不全,并可能在儿童或青少年时期导致终末期肾病(end stage renal disease,ESRD),部分患儿还伴有釉质发育不全,其他症状包括血尿、尿路感染、多尿、多汗、少尿、不完全性远端肾小管酸中毒、高尿酸血症、发育不良等[1,4-5]。目前尚未在CLDN16和CLDN19突变患儿中发现肾脏表型差异的报道。但有研究结果显示,CLDN19基因突变所致的FHHNC患儿出现ESRD的风险是CLDN16基因突变患儿的2倍[6];携带CLDN16基因功能完全丧失突变的患者比携带CLDN16基因部分功能丧失突变的患者更易发生早期疾病和ESRD[7]。同时,因CLDN19在视网膜上皮中也有表达,CLDN19突变患者可伴随严重的眼部受累,包括黄斑疣、色素性视网膜炎、眼球震颤或视力丧失等,而在一些CLDN16突变患者中只有轻微的非特异性眼部受累,如近视、散光、远视或斜视等[8]。
目前,国内外已报道了CLDN16基因的57种致病突变,大多为错义突变,c.453G>T(p.L151F)是CLDN16基因的热点突变,其次为无义突变、移码突变及片段缺失[1]。我国已报道5例由CLND16基因突变导致的FHHNC,1例由CLND19基因突变导致的FHHNC[1,4-5,9-10]。本研究分析了1例维吾尔族近亲家庭CLND16基因C.647G>A纯合错义突变导致的FHHNC患儿。本例患儿以严重佝偻病为主要表现。KARI等[11]报道的7例阿拉伯患儿中有2例伴佝偻病症状。SIKORA等[12]报道的25例FHHNC患者中仅有2例伴佝偻病症状。目前我国报道的患儿中有3例伴佝偻病症状:1个近亲家庭的1对兄妹均为CLND16基因c.574_589delins CCGGTCTGGCT GGACCA片段缺失导致的FHHNC,1例为CLND19基因c.241C>T纯合错义突变[4-5]。DEEB等[13]报道的1个阿拉伯近亲家庭11个孩子中有6个孩子均为CLDN16基因同一位点(C.647G>A纯合)突变导致的FHHNC,1个孩子为杂合突变,均在20岁前出现临床症状,但无佝偻病或其他骨骼系统的异常表现,除最小的男婴(17个月)外,其他患儿均有不同程度的肾功能损伤。本例患儿患有肾结石,肾功能正常,但因FHHNC有显著的ESRD风险,预后差,故患儿肾功能不全的进展情况还需长期随访。DEEB等[13]报道的7例CLDN16基因C.647G>A突变导致的FHHNC患儿及本例患儿的临床表型见表1。

表1 CLDN16基因C.647G>A突变导致的FHHNC患儿临床表型回顾
FHHNC患者伴佝偻病并不常见,且不是主要表现。本例患儿CLND16基因C.647G>A纯合错义突变导致的临床表现与既往报道的同位点突变导致的临床表现有一定差异。有研究结果显示,继发性甲状旁腺功能亢进、代谢性酸中毒和肾功能损伤可能是导致CLDN16突变患者佝偻病发生的部分原因[4,14]。在慢性代谢性酸中毒时claudin-16表达可降低,进而形成离子运输孔道,这可能是慢性代谢性酸中毒患者减慢骨质流失的代偿机制的一部分[15]。同时,由于FHHNC患者骨骼对矿物质流失的适应性反应被破坏,因此骨脱矿增加可能是佝偻病表型形成的原因之一[16]。动物实验结果显示,敲除小鼠CLDN16基因后,小鼠钙、镁的排泄增加,骨密度降低,表现出与人类FHHNC相似的症状[17]。因此,CLDN16基因突变或许是本例患儿出现佝偻病表型的原因之一,确切机制还有待进一步研究。本例患儿在就诊过程中并未出现代谢性酸中毒和肾功能损伤,推测本例患儿出现严重佝偻病表现的病因有:(1)血清钙水平明显低于文献报道[13]的同位点基因突变的患者;(2)镁降低本身可能会抑制甲状旁腺激素分泌,但在FHHNC患者中,严重的低钙血症会引起继发性甲状旁腺激素水平升高;(3)本地区冬季持续时间长,光照不足,患儿未能及时补充维生素D。
由于本例患儿的临床表现与佝偻病相似,因此临床曾初步诊断为佝偻病,并推测病因,包括缺乏维生素D和遗传性低磷性抗维生素D佝偻病,但这些疾病的尿钙排泄量较低,尿磷排出量增加,血清钙正常或稍低,与本例患儿的实验室检测结果不一致。与肾钙质沉着相关的隐性遗传病包括原发性高草酸尿症(有严重的高草酸尿症)、Dent病(有蛋白尿)、Batter综合征(可出现低钾性碱中毒,无双肾钙质沉着)等[1,4-5]。还应注意的是将FHHNC与家族性低镁血症伴继发性低钙血症(TRPM6基因突变)、常染色体显性低镁血症(FXYD2基因突变)等区别开,以上疾病均有低镁血症表现,全外显子基因检测可协助明确诊断。
目前,对于FHHNC主要是对症治疗。因FHHNC有显著的ESRD风险,预后较差,因此疾病的管理尤为重要:(1)密切监测患者的肾功能及电解质水平,应用噻嗪类利尿剂以有效减少FHHNC患者的钙排泄[1];(2)不论是否出现佝偻病症状,均应当适量补充镁剂、钙剂和活性维生素D,以延缓疾病进展,降低甲状旁腺激素水平;(3)出现尿路感染、尿路梗阻、结石等症状应及时治疗;(4)已有研究表明,对于已进展为ESRD的FHHNC患者,肾移植术可使患者的血镁和尿钙调节恢复正常,且不会复发[4,10]。
综上所述,FHHNC的发病率低,但有典型的低镁血症、高钙尿症、肾钙质沉着三联征。本例患儿以佝偻病和手足搐搦症为首发症状,较为少见。本研究通过对同一基因突变位点、不同种族患者临床表现的异质性对比,对FHHNC有了较全面的认识。
