基于基因检测诊断晚婴型神经元蜡样脂褐质沉积症1例
2020-11-04陈星星雷晓燕孙永红胡筱霞赵淑珍
陈星星,雷晓燕,孙永红,胡筱霞,赵淑珍
(甘肃省人民医院 儿科,甘肃 兰州 730000)
神经元蜡样脂褐质沉积症(neuronal ceroid lipofuscinoses, NCLs)是一组遗传异质性溶酶体贮积病,主要表现包括多种类型的癫痫发作、肌阵挛、智力运动发育落后或倒退以及视力损害[1]。主要病因为溶酶体蛋白酶的基因缺陷及结构蛋白的功能失调,导致神经细胞、皮肤上皮细胞、肌细胞和淋巴细胞内脂褐素沉积[1-2]。目前报道的发病率为(0.1~7.0)/10 万,不同地区会有差异[3],国内报道较少。NCLs最初根据发病年龄、临床表现以及电镜下沉积物特征在临床上可分为婴儿(Haltia-Santavuori)、晚期婴儿(Jansky-Bielschowsky)、青少年(Batten,Spielmeyer-Vogt)和成人(Kufs)共4种亚型,而现今根据基因型,NCLs可分为CLN1~CLN14共14种亚型,其中CLN2和CLN3型较为常见[1-4]。
不同亚型的NCLs疾病根据发病年龄不同可有不同表现[1, 4-5],其中CLN2为经典的晚期婴儿型:2~4岁间发病,始发通常为癫痫发作和(或)共济失调,随后可出现获得性里程碑式发育落后,难治性癫痫,共济失调或进一步恶化,以及锥体征束表现。发作可表现为各种类型的肌阵挛发作、非典型失神发作、强直-阵挛发作。视觉障碍通常出现在4~6岁,并且进展迅速,最终视力丧失,大多数CLN2确诊患者在卧床不起和失明后过早死亡,预期寿命从6岁到十几岁。CLN2型疾病为常染色体隐性遗传疾病,由TPP1基因突变所致,此基因编码蛋白为三肽基肽酶,其可从蛋白质的N末端顺序去除三肽[5]。该报道对一例结合临床表型、全外显子组测序技术(whole exome sequencing, WES)诊断的CLN2进行讨论和研究,通过相关基因诊断技术和遗传学分析对这类临床表型隐匿,分型复杂的疾病进行再次认识。
1 资料与方法
1.1病历摘要 患儿,男,4岁,主因“诊断癫痫20月,发现运动障碍伴语言发育倒退8个月”于2018-12-23入院。3岁1个月时出现间断抽搐,4岁1个月开始出现运动、语言倒退、肢体抖动,5岁开始出现肢体抖动加重、视力下降、双眼感光消失、失语、瘫痪在床。
1.2研究方法
1.2.1临床诊断与家系分析 收集患者家族史、临床表现、实验室检查等临床资料。
1.2.2样本采集与测序实验 在患者及其父母知情同意的情况下,采取患者及其父母的外周血样本,使用血液基因组提取试剂盒(UnigeneDx)进行DNA提取,并用Life Technologies 公司生产的Qubit测定DNA的浓度进行定量。参照KAPA HyperPlus(KAPA Biosystem, Roche)文库准备试剂盒及SeqCapEZ捕获试剂盒(NimbleGen, Roche)对基因组DNA进行片段化、末端修复、接头连接及杂合捕获,制备外显子捕获文库。文库采用HiSeq X Ten (Illumina)测序平台进行双端测序。该研究获得甘肃省人民医院伦理委员会的审查批准。
1.2.3数据分析及验证 采用FASTQC(v0.11.5)对测序得到的原始数据进行质量评估,通过BWA软件(v0.7.15)[6]工具将测序得到的.fastq文件比对到参考基因组上(基因组版本为hg19),利用Samtools(v1.3.1)、Picard、GATK等工具对比对结果进行格式转化、排序、去重、重排以及碱基质量矫正,利用Annovar工具对突变位点进行注释[7-8]。最后根据美国医学遗传学和基因组学会(ACMG)分类指南并结合患者癫痫的临床表型进行相关变异的筛选[9]。变异分析过程中参考公共人群频率数据库(ExAC、ESP、1000Genomes及gnomAD)与dbSNP、OMIM、HGMD、ClinVar等多种数据库进行变异的致病性评估。根据分析所得的候选变异位点,在NCBI网站下载TPP1序列并设计聚合酶链式反应(polymerase chain reaction,PCR)引物,对患者及其妹妹和父母的DNA 样本进行PCR扩增后,通过Sanger测序进行序列的比对验证。
1.2.4突变致病性预测与蛋白序列同源性比对 利用蛋白功能预测软件(SIFT、Polyphen2及MutationTaster)对测序数据所得变异的致病性进行预测。通过Uniport及Ensembl数据库查找并下载人及其他物种的TPP1蛋白序列,使用Jalview软件进行蛋白同源序列比对[10]。
2 结 果
2.1临床诊断与家系分析 患儿出生时视力正常,3岁1个月时出现间断抽搐,曾就诊于某三甲医院,完善脑电图、头颅MRI等相关检查后确诊为癫痫,给予丙戊酸钠口服治疗,患儿抽搐有所好转。于4岁1个月时家长发现患儿运动逐渐倒退,具体表现为不能独自行走,走路不稳,正常行走不超过20 min,伴四肢不自主抖动,出现语言发育倒退,只能叫“妈妈”,且吐字不清,考虑运动障碍性脑病,且8个月来上述症状进行加重。入院体格检查:意识清楚,精神尚可,呼吸平稳,面色尚可,无特殊面容,构音不清,言语笨拙,双瞳孔等大、正圆,直径约3 mm,对光反应灵敏,眼球运动充分,未见眼球震颤,颈软无抵抗感,心肺腹查体未见阳性体征,四肢及关节无畸形,双下肢肌力Ⅳ级,肌张力正常,走路不稳,伴躯干及四肢不自主抖动,双下肢无水肿,神经系统查体双巴氏征阴性;双膝腱反射、跟腱反射、轮替试验、指鼻试验、跟膝胫等试验不能配合检查。辅助检查:血常规、生化全套、血乳酸、血氨、同型半胱氨酸、铜蓝蛋白、尿常规均正常。丙戊酸钠血药浓度在正常范围。颈部血管超声:双侧颈动脉、颈静脉超声未见明显异常。动态视频脑电图示:异常幼儿脑电图(背景活动偏慢,睡眠期双侧额、颞区为主多灶性棘波、棘慢波、尖波及慢波发放)。颅脑MRI示:双侧大、小脑半球脑萎缩改变,请结合临床;右侧上黏膜增厚(图1)。患儿于4岁11月时开始出现视力下降,视物不清,视野逐渐减小,至5岁时,双眼感光完全消失,肢体不自主抖动加重,四肢无力,瘫痪在床,伴失语。该患者的发病年龄在4岁左右,视力进行性减退,临床症状以癫痫,运动、语言倒退伴视力丧失为主。
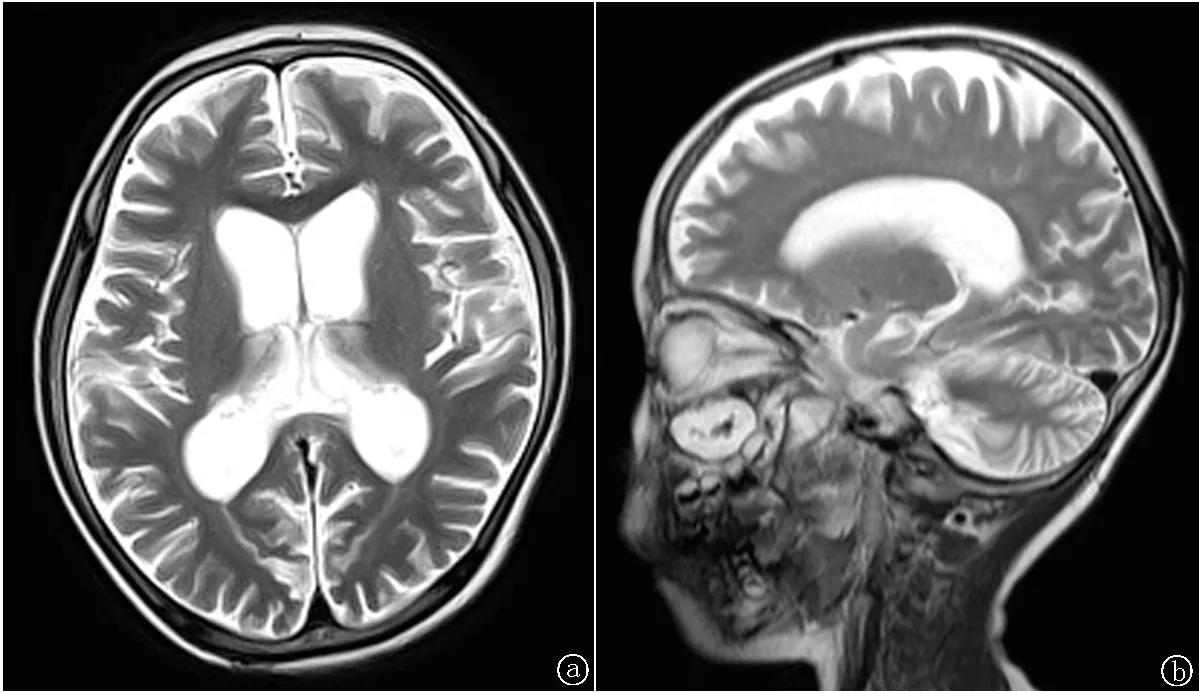
图1 双侧大、小脑半球脑萎缩改变
患者父母均未出现类似症状,家族中未出现类似病史。结合患者临床表现、动态视频脑电图、头颅MRI、基因检测等相关检查,诊断 JNCL,CLN2 型(晚婴型)。
2.2全外显子组测序与 Sanger 测序验证 本次在受检者TPP1 基因上检出两个突变位点:已报道的致病位点c.1424C>T(p.Ser475Leu)和 新发突变位点c.962A>G(p.His321Arg) 。患者父亲样本检出 TPP1基因上c.1424C>T(p.Ser475Leu)(图2)杂合变异;患者母亲样本检出TPP1基因上c.962A>G(p.His321Arg)杂合变异(图3)。一代测序证实,先证者的c.1424C>T 变异遗传自父亲,c.962A>G 变异遗传自母亲,这两变异在染色体上为反式排列。TPP1基因突变会导致神经元蜡样脂褐质沉积症2型,遵循常染色体隐性遗传。并对以上2个突变位点进行 Sanger 测序验证,证明确实存在。

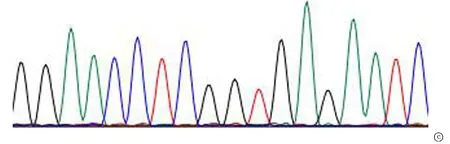
图2 TPP1 c.1424 位点Sanger测序图 TPP1基因c.1424C>T(p.Ser475Leu) 突变遗传来自父亲。 a.患者TPP1基因上检测到c.1424C>T(p.Ser475Leu)突变;b.患者父亲TPP1基因上检测到c.1424C>T(p.Ser475Leu)突变; c.患者母亲TPP1基因上未检测到c.1424C>T(p.Ser475Leu)突变
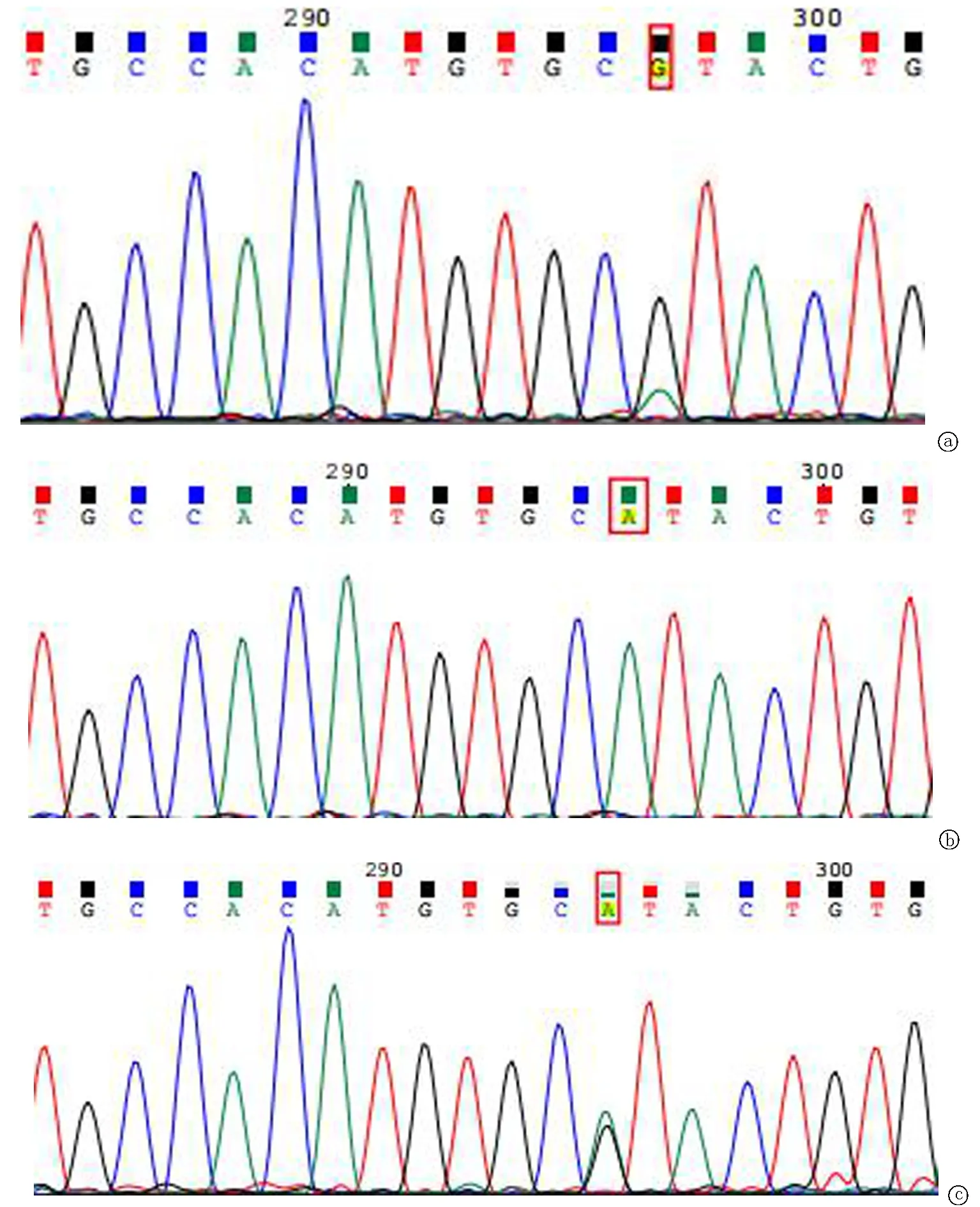
图3 TPP1 c. 962位点Sanger测序图 TPP1基因c.962A>G(p.His321Arg) 突变遗传来自母亲。a患者 TPP1基因上检测到 c.962A>G(p.His321Arg)突变;b患者父亲TPP1基因上未检测到c.1424C>T(p.Ser475Leu)突变;c患者母亲TPP1基因上检测到 c.962A>G(p.His321Arg) 突变
2.3突变致病性预测分析 突变位点1 c.1424C>T(p.Ser475Leu):①表示TPP1基因CDS区的第1424号核苷酸由C突变为T,导致编码的第475号氨基酸由丝氨酸突变为亮氨酸;②此变异在普通人群中的最大携带频率为0.039%,提示变异在普通人群中罕见(PM2);③ 此变异位于肽酶 S8/S53 功能结构域(PM1); ④ 此变异与已报道的另一致病变异 c.509-1G>C曾在CLN2患者中有检出,两者在染色体上为反式排列[11](PM3);⑤ 多个软件(SIFT,MutationTaster,PolyPhen2)预测此变异有害(PP3); ⑥此变异在 Clinvar 和 LOVD 数据库中有多次提交记录,其中至少有 2 例为疑似致病变异记录; ⑦ 一代测序证实,先证者的父亲也携带有此变异,先证者的母亲和妹妹均未携带此变异,证实先证者的此变异遗传自父亲;⑧TPP1 基因编码三肽基肽酶,其突变会导致CLN2,遵循常染色体隐性遗传;⑨该例受检者表型与 CLN2 疾病部分符合。根据ACMG指南(PM2+PM1+PM3+PP3),暂将 c.1424C>T(p.Ser475Leu)变异判为疑似致病变异。
突变位点2 c.962A>G(p.His321Arg):①此变异表示 TPP1 基因 CDS 区域的第 962 号核苷酸由 A 突变为 G,导致编码的第 321 号氨基酸由组氨酸突变为精氨酸;②此变异在多个普通人群数据库(1000g、ExAC 及 gnomAD)中未见报道,提示变异在普通人群数据库中极为罕见(PM2);③多个软件(SIFT,MutationTaster,PolyPhen2)预测此变异有害(PP3);④此变异位于肽酶 S8/S53 功能结构域(PM1);⑤暂未发现此变异存在相关临床或功能报道。一代测序结果显示,先证者的母亲携带有此变异,父亲未携带,证实先证者的此变异遗传自母亲。此变异与本次检出的另一疑似致病变异(c.1424C>T)在染色体上为反式排列,符合 CLN2 疾病的常染色体隐性遗传模式。综上,根据 ACMG 指南(PM2+PM1+PP3+PM3),暂将 c.962A>G(p.His321Arg)变异归为疑似致病变异。
2.4蛋白序列同源性比对 以上两个突变位点,在人类和其他物种同源蛋白相同区域高度保守不易发生改变,TPP1表达蛋白第 475 位氨基酸基本为 Ser,第 321 位氨基酸基本为His(图4)。
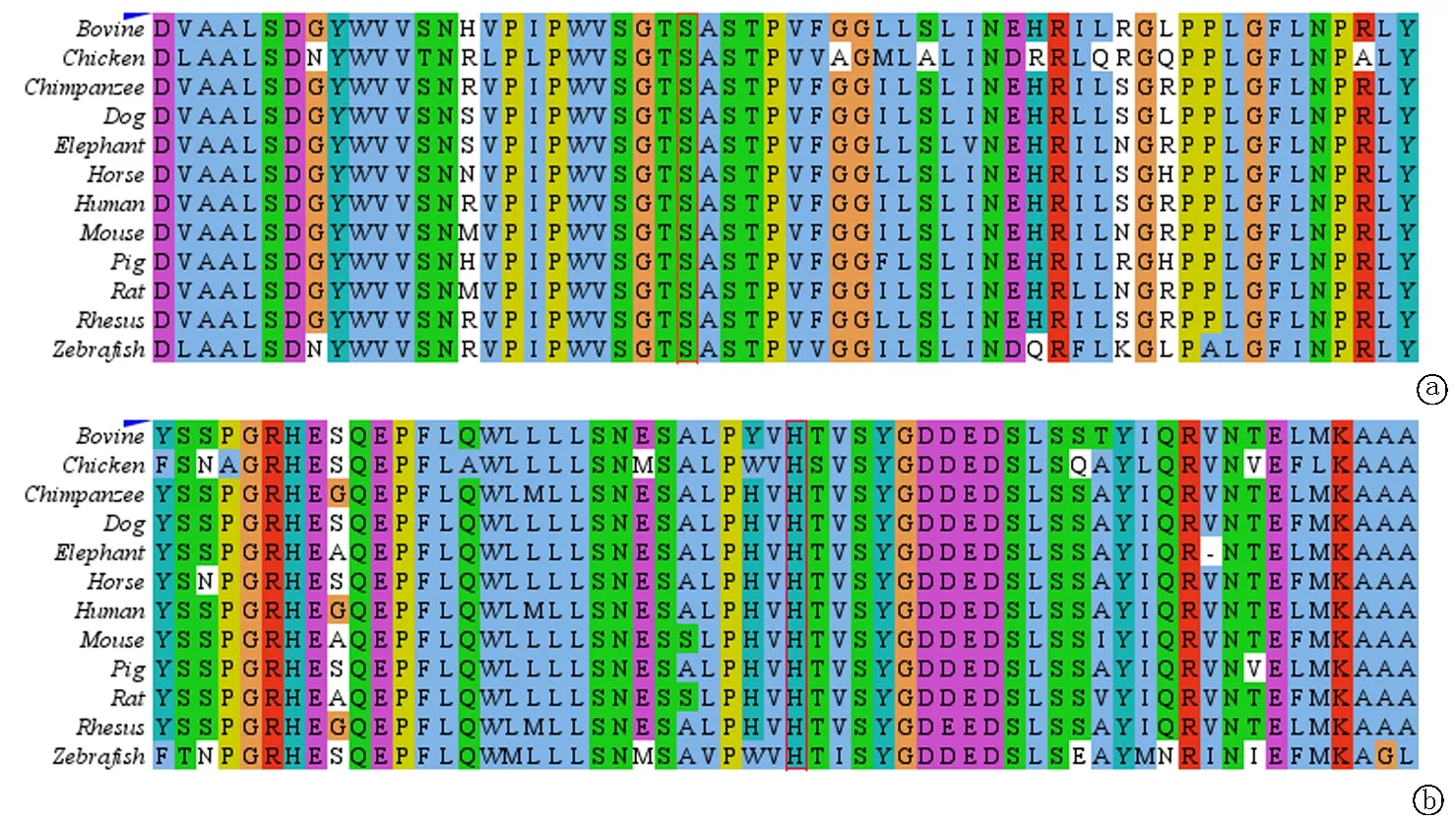
图4 TPP1 c.1424C>T(p.Ser475Leu)和TPP1 c.962A>G(p.His321 Arg) 蛋白序列同源性比对 a.TPP1 c.1424C>T(p.Ser475Leu)蛋白同源性比对(红色框标注的为变异位点,结果显示此位点在多个物种中保守);b.TPP1 c.962A>G(p.His321 Arg)蛋白同源性比对(红色框标注的为变异位点,结果显示此位点在多个物种中保守)
3 讨 论
NCLs是一组遗传异质性溶酶体贮积病,其临床表现复杂,诊断主要通过临床表现、病理、脑电图、 影像学及分子生物学确诊,由于诊疗水平有限,该病极易误诊。文中受检者单凭基因检测结果不能确诊,但综合临床表现(与CLN2部分符合)、头颅MRI结果、脑电图资料及基因检测结果最终诊断为CLN2。现根据基因型可分为CLN1~CLN14共14种亚型,其中CLN2疾病为经典的晚期婴儿型[1]。
CLN2遵循常染色体隐性遗传,可由TPP1/CLN2突变导致其编码的三肽基肽酶失活而引起CLN2的一系列临床表现[5, 12],该病尚无特效治疗方法。国内报道的NCLs患者突变基因主要是 CLN1、CLN2、CLN3、CLN5、CLN6、CLN7,临床多表现为难治性癫痫、视力下降、智力下降、精神及运动功能障碍、性格行为改变、记忆力下降,且在中国人群NCLs病例报道中,婴儿型(INCLs)13例(12.9%),晚期婴儿型(LINCLs)48 例(47.5%),青少年型(Batten 型,JNCLs) 30例(29.7%),成年型(ANCLs)10例(9.9%);其中仅32例完善了基因检测[13]。
由于大多数 CLN2 确诊患者在卧床不起和(或)失明后过早死亡,然而,不可逆的神经退行性变通常发生在5岁确诊之前[14],最近批准的脑室内酶替代疗法已被证明可以有效减缓CLN2疾病患者运动和语言功能的快速下降[15],因此早期诊断对优化护理、改善患者生活质量就显得更加重要。但是在我国,由于对该疾病认识低、临床表现不明确以及诊断检测的可及性有限,诊断延迟是常见的。但是在诊断方面,除了基因检测,还有头颅MRI、脑电图均可提示CLN2疾病的诊断,例如,严重的小脑萎缩是MRI诊断时的主要表现[16],脑电图检测到的光敏性也是CLN2疾病的早期标志[17]。
CLN2的主要病因为TPP1/CLN2基因突变引起溶酶体蛋白酶的基因缺陷及结构蛋白的功能失调,导致神经细胞、皮肤上皮细胞、肌细胞和淋巴细胞内脂褐素沉积[5]。目前已报道了的TPP1超过 100 种突变,包括错义突变、无义突变、剪接变异、插入缺失等,突变分布整个编码区[12],但是目前TPP1在体内的底物尚不清楚,TPP1酶缺乏症的分子、病理机制尚不清楚[18-20]。TPP1/CLN2基因位于11p15染色体上[21],包含13个外显子,长度为6.7 kb,编码溶酶体外肽酶(TPP1),酸化后以无活性的原酶形式被加工成一个46 000的蛋白质。成熟的酶从溶酶体降解的小多肽的氨基末端裂解三肽,并且具有弱的内肽酶活性[22]。
迄今为止,在389名TPP1缺乏症患者中报告了131个TPP1基因变异,在CLN2疾病中,大多数患者(60%)有两种常见的变异之一(c.509-1 G>C和C.622 C>T[p.(Arg208*)]),这两个变异使密码子过早终止,一直被报道有致病性[13]。如果患者有任何CLN2疾病的迹象,并且分子检测发现TPP1中有任何致病或可能致病的变异,TPP1酶活性检测可用于确诊[5]。但是,在本例报道中由于实验条件所限,尚未进行TPP1酶活性的检测,这是本文的不足之处。
CLN2为常染色体隐性遗传,即等位基因上有2个有害突变可能导致疾病的发生。 本文检测到的TPP1: c.1424C>T(p.Ser475Leu)和c.962A>G(p.His321Arg) 在正常人群中发生概率极低。 TPP1 基因 CDS 区的第 1424 号核苷酸由 C 突变为 T,导致编码的第 475 号氨基酸由丝氨酸突变为亮氨酸,此变异与已报道的另一致病变异 c.509-1G>C 曾在CLN2患者中有检出,两者在染色体上为反式排列[11]。此外,另一个突变位点c.962A>G(p.His321Arg),提示TPP1 基因 CDS 区域的第 962 号核苷酸由 A 突变为 G,导致编码的第 321 号氨基酸由组氨酸突变为精氨酸,此变异在多个普通人群数据库(1000g、ExAC 及 gnomAD)中未见报道,提示变异在普通人群数据库中极为罕见;此变异位于肽酶 S8/S53 功能结构域,表明该突变位点对TPP1蛋白的正常功能至关重要,目前暂未发现此变异存在相关临床或功能报道。一代测序结果显示,先证者的母亲携带有此变异,父亲未携带,证实先证者的此变异遗传自母亲。此变异与本次检出的另一疑似致病变异(c.1424C>T)在染色体上为反式排列,符合 CLN2 疾病的常染色体隐性遗传模式。以上两个突变位点,在人类和其他物种同源蛋白相同区域高度保守不易发生改变,对行使蛋白质的正常功能具有重要意义。以上结果均支持 c.962A>G(p.His321Arg)可能为 CLN2 疾病新发现的致病突变。综合以上遗传学分析得出,TPP1 的复合杂合突变极有可能导致晚婴型NCLs。
新生儿筛查是改善目前可治疗疾病的早期诊断的最有效方法,有专家建议,用与串联质谱检测相兼容的酶底物测定TPP1活性,可支持未来大规模的NBS计划[5, 23]。但是新生儿筛查CLN2项目的成功主要依赖于基因型-表型相关性的清晰性,因此,我们必须共同努力确定TPP1变异所导致的疾病可能性,以便通过人群筛选和诊断分子遗传检测来解释检测到的变异。随着分子遗传检测作为小儿起病神经系统疾病的一线诊断检测日益增多,全面的报告和数据共享至关重要。临床上发现TPP1/CLN2突变携带者,下一代致病概率较高,建议进行早期遗传咨询及家系验证。本研究可为该家系遗传咨询及再生育指导提供依据,新突变基因的发现也扩大了该基因的突变谱,为阐明 NCLs 遗传机制提供线索和方向。
