汉族与蒙古族肠道菌群结构差异分析
2020-10-31张沐诗毛明慧谢丹肖瑶李树春
张沐诗 毛明慧 谢丹 肖瑶 李树春
中央民族大学药学院民族医药教育部重点实验室(北京100081)
大多数肠道微生物对人体生理和健康都有着重要影响,它们对人类生命至关重要[1]。肠道菌群与肠道细胞之间的相互作用可以调节屏障功能,不断刺激免疫系统防御病原体[2]。肠道菌群与宿主之间的平衡如果发生了改变,可能会导致机体产生各种疾病[3]。
我国是一个多民族国家,少数民族人群存在聚集生活的状态,且各民族间的肠道菌群存在差异[4]。蒙古族作为我国北部少数民族之一,饮食习惯、地理环境、社会文化等有自己独特的地方,且纯系人口较多,适合作为民族差异研究的对象。最近研究表明各民族之间疾病的患病率也存在着种族和民族差异[5-6]。已有研究表明,汉族和蒙古族糖尿病患病率存在差异[7]。孙刚等[8]在比较内蒙古地区蒙古族和汉族高血压患病率及危险因素分布差异时,证实内蒙古地区蒙古族人群高血压患病率高于汉族。另一项中国少数民族研究显示[9],在汉族和蒙古族之间,人乳头瘤病毒(HPV)患病率存在明显的种族差异,汉族患病率(11.5%)显著低于蒙古族(32.6%)。因此,种族、微生物群和健康差异之间的相互联系非常有研究意义。鉴于此,为了探讨汉族和蒙古族肠道菌群差异,本论文通过高通量测序和生物信息学分析寻找汉族或蒙古族特有菌群,为从民族角度分析肠道菌群和疾病关系研究提供新的研究思路和实验支持。
1 资料与方法
1.1 样本和信息采集本研究的开展经过了中央民族大学伦理委员会的审核(批准号:ECMUC 2019003CO),并让每个志愿者都签存了知情同意书。本研究的样本采集地点为蒙古阿拉善盟阿左旗,受试者主要来自当地人群,一共获得粪便样本28 例,样本采集后30 min 内用粪便储存液保存,干冰运输到实验室后,置于-80 ℃冰箱中冷冻保存。研究对象的纳入标准:汉族志愿者三代直系血亲均为汉族人群,蒙古族志愿者三代直系血亲均为蒙古族人群;所有志愿者都遵循其传统的生活习惯。研究对象的排除标准:患有急性或慢性炎症性疾病;患有慢性胃肠疾病;在采集样本前三个月内使用过抗生素或益生菌;三个月内使用过激素类药物;正在哺乳的女性。
1.2 粪便菌群DNA 提取粪便菌群基因组DNA提取采用PowerSoil® DNA Isolation Kit,具体操作按其说明书进行。PCR 扩增体系为:Phusion Master Mix(2×)15 μL,上游引物3 μL,下游引物3 μL,基因组DNA 10 ng,无菌水2 μL;PCR扩增条件为:98 ℃预变性1 min;30 个循环包括(98 ℃,10 s;50 ℃,30 s;72 ℃,30 s);72 ℃,5 min。使用提取出的DNA 作为PCR 扩增模板,测序区域为V3+V4 区,使用特定带Barcode 的引物。将PCR 产物稀释成同样浓度,再将产物混匀。使用琼脂糖凝胶电泳的方法纯化产物,凝胶的配制为1×TAE 浓度的2%琼脂糖凝胶。根据主带大小,选择在400~450 bp的条带,并割胶回收,用于Illumina 制备试剂盒进行构建文库,测序使用HiSeq2500。
1.3 生物信息分析及统计分析使用QIIME软件[10]对原始序列进行过滤,舍弃低质量的原始序列,得到Clean tags,并构建α稀释曲线。基于数据库比对,使用USEARCH[11]舍弃序列中Barcode 文件,获得可以用于后续分析的Final tags。USEARCH构建进化树,生成OTU 丰度表和代表性序列。在QIIME 中生成Alpha 多样性表,并利用R 语言ggplot2 包作盒形图。使用未加权unifrac 算法和加权unifrac 生成β-多样性,用来研究物种分布是否受到民族或疾病因素的影响,显著性用adonis 方法检验。维恩图用于统计汉族和蒙古族差异OTU 的交并集情况。使用RDP classifier 网页的注释文件对OTU 进行物种注释,多数OTU 可以注释至属水平。使用KRONA[12]对汉族和蒙古族群体各层次的物种比例进行可视化显示。
通过PICRUSt[13](http://PICRUSt.github.io/PICRUSt/)分别对汉族和蒙古族样本进行功能和代谢途径预测分析,利用QIIME 获得Greengenes 数据库注释的OUT 表,与KEGG 数据库进行比对,获得功能预测信息。利用STAMP 软件在P<0.05 显著性水平对预测结果进行分析,获得具有显著性差异的代谢通路信息,并对结果进行可视化。
1.4 统计学方法所有样本的数据均表示为均值±标准差,方差齐性的数据进行F检验,如果不满足方差齐性,采用校正的t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 研究对象的临床特征本研究最终纳入10例汉族样本,18例蒙古族样本。汉族组平均年龄(52.50±8.56)岁,蒙古族组平均年龄(56.88±14.27)岁。结果显示汉族与蒙古族两民族之间差异无统计学意义(P>0.05),见表1。
2.2 微生物多样性10 份汉族粪便样本,每个样本平均有47 391条Final tags;18份蒙古族粪便样本,每个样本平均有48 433 条Final tags。测序质量在QIIME 软件中生成Rank-Abundance 趋向水平直线,表明测序的数据量足够大,因此测序结果可用于生物多样性分析和统计分析。Alpha 多样性即样品内部的生物多样性,本研究根据OTU table 和系统发育树计算参数,包括chaol 指数、Shannon 指数。本研究的α多样性分析经Wilcoxon 秩和检验结果显示,Chao1 和Shannon 指数在两民族无显著差异(P(Chao1)=0.87,P(Shannon)=0.58)。在R 语言中基于非加权距离矩阵(un)weighted_unifrac 作图,组间差异使用adonis 检验,当仅纳入菌群多样性作为参考时(距离为非加权矩阵),汉族和蒙古族菌群之间差异有统计学意义(P=0.001);而同时把菌群种类和数量同时纳入参考时(距离为加权矩阵),汉族人肠道菌群和蒙古族之间差异有统计学意义(P=0.001),见图1。
2.3 微生物组成分析每个OTU 代表不同的微生物物种,Venn 图可以表示两种族群肠道微生物的多样性情况。结果表明,汉族肠道菌群共分析出有1 011 个OTU,其中特有OTU 446 个,且特有的OTU 数量约为蒙古族的两倍。蒙古族肠道菌群总共分析出有784 个OTU,特有OTU 219 个。两组合起来分析出OTU1230 个,共有的OTU 为565 个,占总体的45.9%(图2)。

图1 微生物多样性分析Fig.1 Microbial diversity analysis
使用R 语言中edgeR 包作汉族和蒙古族肠道菌群差异分析,其原理是基于负二项分布模型,为消除测序产生的误差和聚类时产生的噪音,在用edgeR 作组间差异分析时,删除丰度<0.001 的OTU,对OTU 表中的丰度信息进行过滤,即总体样本中某个OTU 的丰度值小于总体OTU 丰度值的千分之一(P<0.05),则舍弃。本实验共分析OTU 1 245 个,从火山图知,汉族和蒙古族组间有309个差异OTU。注释到科水平的OTUs 包括拟杆菌科(bacteroidaceae)、克里斯滕内尔科(christensenellaceae)、肠杆菌科(enterobacteriaceae)、erysipelotrichaceae、毛螺菌科(lachnospiraceae)、紫单胞菌科(porphyromonadaceae)、红螺科普雷维菌科(prevotellaceae rhodospirillaceae)、瘤胃菌科(ruminococcaceae)、韦荣球菌科(veillonellaceae)。相较于汉族,蒙古族OTUs 丰度显著增加的有170 个,显著下调的有139 个(图5),火山图证实本实验纳入的汉蒙两组人群肠道菌群存在差异。
2.4 物种注释及菌群分布在USEARCH中对原始测序文件Final tags1 和Final tags2 进行拼接,拼接后过滤、去重,运行cluster_otus 脚本聚类并去除嵌合体,运行align_seqs.py 脚本构建进化树,然后以默认的97%相似度进行OTUs 聚类分析,最后生成OTU 表。下载RDP classifier 网页的注释文件16S rRNA training set 16,将生成OUT 与之进行比对,进行注释。利用Krona 软件将注释后的OUT表可视化,展示结果中,圆圈从内到外依次代表不同的分类级别,扇形的大小代表不同OTUs 注释结果的相对比例。门水平上,汉族中丰度最高的是厚壁菌门(firmicutes)占比52%,其次是拟杆菌门(bacteroidetes)(36%)、变形杆菌门(proteobacteria)(9%)、放线菌门(actinobacteria)(3%)。蒙古族人群在门水平上,丰度最高的四个门是厚壁菌门(firmicutes)(53%)、拟杆菌门(bacteroidetes)(42%)、放线菌门(3%)(actinobacteria)、疣微菌门(verrucomicrobia)(0.75%)。见图3。
在属水平上,蒙古族和汉族丰度最高的前四个是拟杆菌属(Bacteroides)、粪杆菌属(Faecalibacterium)、普雷沃菌属(Prevotella)、狭义梭菌属(Clostridium sensu stricto)。此外,检验结果显示在蒙古族人粪便中丰度含量大于1%且显著(P<0.05)高于汉族组的细菌属有:梭菌Ⅳ(ClostridiumⅣ)、副杆状菌属(Parabacteroides)、枝菌属(Alistipes),在汉族组中丰度含量大于1%且显著(P<0.05)大于蒙古族组的细菌属有:瘤胃球菌属(Ruminococcus)、克雷白氏杆菌(Klebsiella)、布劳特氏菌属(Blautia)、毛螺旋菌属(Parasutterella)。综上所述,两种族肠道微生物中优势菌门和菌属相同,但其他细菌属的丰度差异较为明显。

图2 微生物组成分析Fig.2 Microbial composition analysis
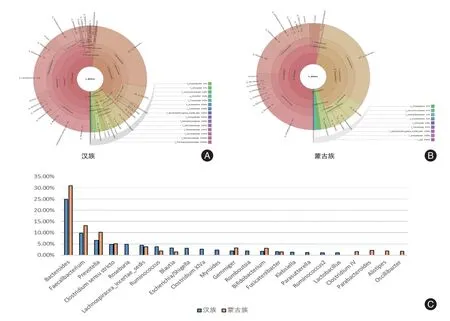
图3 物种注释及菌群分布Fig.3 Species annotation and flora distribution
2.5 PICRUSt 功能预测分析使用QIIME 软件运行cluster_otus 命令对测序获得的16S rRNA 基因序列进行OUT 聚类,得到BIOM 格式的OTU table;运行single_rarefaction.py 对OTU 表进行校正,得到每个OTU 更加真实的丰度;使用predict_traits.py 命令映射、推断、量化代谢功能,得到两民族间肠道菌群代谢功能差异,使用STAMP 软件对结果进行可视化展示。通过PICRUSt 分别对汉族和蒙古族样本进行肠道菌群的功能和代谢途径预测分析,结果发现汉族与蒙古族之间共有11 个代谢通路有显著差异,其中蒙古族显著高于汉族的代谢通路有10 个,包括N- 糖链的生物合成代谢通路、醚脂代谢通路、异喹啉生物碱的生物合成通路等。汉族显著高于蒙古族的代谢通路只有1 个为胰岛素信号通路(图4)。

图4 汉族和蒙古族肠道菌群功能预测分析Fig.4 Functional prediction analysis of intestinal microflora of Han and Mongolian
3 讨论
许多常见疾病与肠道微生物的组成和种族有关,这提出了一个核心假设,即种族之间的肠道微菌群差异是否可以反应出种族之间的体质差异。中国的蒙古族和汉族在社会经济,文化,地理,饮食和遗传多样性有很大不同,并且个体差异和环境因素也会影响整个微生物群的组成。为了证实种族-微生物群假说,我们评估了蒙古族与汉族健康个体中总肠道微生物群的差异。最后得出结论,蒙古族和汉族之间肠道菌群存在较大差异,并找到了差异菌属。
本研究从蒙古族和汉族两个数据集的结果中发现,汉族人和蒙古族人肠道细菌的优势细菌门和优势菌属相似,优势菌门均为厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes),优势细菌属均为拟杆菌属(Bacteroides)、费氏菌属(Faecalibacterium)、普雷沃菌(Prevotella)、敏感梭菌(Clostridium sensu stricto)。厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)在健康人体肠道菌群中占比最多,已经有多项研究表明了这一现象[14]。拟杆菌属和费氏菌属是肠道中的有益菌。临床前实验表明拟杆菌属被广泛认为是控制新陈代谢、预防癌症和减轻炎症的新靶点,原理是通过调节淋巴细胞和细胞因子的表达[15],而费氏菌属的减少则和人体内炎症性肠病[16]、肥胖[17]、非小细胞肺癌[18]相关。Prevotella 和Clostridium sensu stricto 在肠道中主要参与碳水化合物分解代谢,促进人肠道营养吸收[19]。这四种优势菌属是人体内的主要菌属,在肠道中发挥重要作用。
但基于α、β多样性研究和火山图,发现汉族和蒙古族组间有显著差异,相较于汉族,蒙古族OTU 丰度显著增加的有170 个,丰度显著下调的有139 个,组间共有309 个差异OTU(P<0.05),科水平上的主要差异菌群拟杆菌科、克里斯滕内尔科、肠杆菌科等;注释到属水平有Acetatifactor、鲍曼不动杆菌(Acinetobacter)、放线菌属(Actinomyces)等。先前有研究表明汉族和蒙古族糖尿病患病率存在差异[20],两族人群发病率差别可能主要与饮食习惯等有关,但肠道菌群的差异也可能会影响两组间的疾病发病率。例如双歧杆菌属(Bifidobacterium)在蒙古族肠道菌群中丰度位列第五,其可产生胆碱代谢物,并且可能参与饮食诱导肥胖和糖尿病[21]。蒙古族瘤胃球菌属(Ruminococcus)丰度低于汉族,一些饮食碳水化合物可能会直接有利于产生丁酸的细菌生长如该菌属[22]。瘤胃球菌属(Ruminococcus)可以帮助宿主参与淀粉的分解,分解淀粉产生的短链脂肪酸和能量对机体有帮助作用[23]。瘤胃球菌属(Ruminococcus)菌群的显著上调可能与两民族间饮食差异有关,蒙古族有以奶食、肉食等高脂蛋白为主的饮食习惯,而汉族人则主要以碳水化合物为主食,因此瘤胃球菌属(Ruminococcus)在汉族人肠道菌群中丰度较高也可能是食物调控的结果。
通过PICRUSt 分别对汉族和蒙古族样本进行功能和代谢途径预测分析,发现汉族显著高于蒙古族的代谢通路为胰岛素信号通路,在此之前,有人研究发现蒙古族糖尿病发病率高于汉族[11],这预示着也许肠道菌群的胰岛素信号通路在正常人的降糖功能中发挥了作用,种族间的肠道微生物差异间接影响着种族的发病率。不同种族肠道中共生菌群差异可能由多种因素引起,包括饮食结构[24],环境暴露,社会文化影响,人类遗传变异[25]等。不管原因如何,导致的差异都最终会影响到人体内代谢过程和体内平衡。
本研究采用的是是基于16S rRNA 基因高通量测序技术对汉族和蒙古族人的肠道菌群进行研究,尽管得到的结果是显著性的,但是仍然需要考虑到研究中的不足之处,比如样本处理和OTU 聚类的变化以及抽样不均等,这些只能通过加大样本量来解决。研究结果强调了在健康人群中两民族肠道菌群的差异性,意味着在将微生物群差异与疾病联系起来的研究中,需要考虑种族差异作为潜在的混杂因素。这些结果的进一步研究方向,可能是将特定的微生物族群作为健康差异的潜在诊断或治疗方法。
