18F-硝基咪唑的放化稳定性
2020-10-28钟新文陈尚东
时 光,钟新文,程 亮,陈尚东
1.吉林警察学院 刑事科学技术系,吉林 长春 130123;2.沈阳化工大学 应用化学学院,辽宁 沈阳 110142
18F-硝基咪唑,化学名1-H-1-(3-18F-2-羟基丙基)-2-硝基咪唑(18F-fluoromisonidazole,18F-FMISO)是一种常见的乏氧硝基咪唑类PET显像剂,与乏氧细胞有较高的亲和力[1],已广泛应用于临床研究。恶性肿瘤的迅速生长依赖于肿瘤血管的生成,肿瘤血管的供养能力差、血流缓慢、不规则和异常弯曲等现象,会引起肿瘤组织氧和营养物质不充分,从而产生肿瘤乏氧现象。文献[2]报道过18F-FMISO的合成方法,氟化后水解再用碱中和反应体系(见图1),与本研究的方法基本相同,差别在于纯化方法及用半制备型HPLC纯化产品流动相的选择。范文博等[3]报道了18F-FMISO的纯化均采用乙醇水溶液作为流动相,所得到产品中一定会含有乙醇成分;早期也有采用固相萃取柱作为纯化手段[4]。本研究拟采用乙腈水溶液做为流动相,分离产品后进入旋转蒸发仪除掉有害溶剂,再用生理盐水溶解产品,以得到不含有乙醇的18F-FMISO溶液。并分别选择抗坏血酸、抗坏血酸钠做为稳定剂,测量产品放化纯度变化,选择出合适的稳定剂。

图1 18F-FMISO合成原理
1 实验部分
1.1 仪器与试剂
HM-10HC回旋加速器和CFN MPS-200型合成装置,日本住友重工业株式会社;高效液相色谱仪(ZORBAX SB-C18纯化柱),美国Agilent公司;放射性薄层色谱仪,美国BIOSCAN公司。
H218O,丰度97%,上海化工研究院;无水乙腈、碳酸钾、抗坏血酸钠、抗坏血酸,美国SIGMA-ALDRICH公司;Kryptofix2.2.2(K2.2.2)、1-(2’-硝基-1’-咪唑基)-2-氧-四氢呋喃基-3-氧-甲苯磺酰基-丙二醇(NITTP)、1-H-1-(3-19F-2-羟基丙基)-2-硝基咪唑(19F-FMISO),德国ABX公司;4-(4-甲基哌啶)吡啶阴离子交换树脂(简称QMA柱),美国Waters公司;可裁硅胶板,德国Macherey-Nagel公司;乙醇、甲醇,色谱纯,北京化学试剂厂。
1.2 实验方法
1.2.118F-的生产 用住友HM-10HC回旋加速器,以60 μA的质子束流连续轰击H218O。H-经高频电场反复加速,最后由碳膜剥离轰击到靶上,发生18O(p, n)18F核反应生产18F-,50 min后用氦气将18F-从靶传到住友CFN MPS-200合成模块的靶水回收瓶中。
1.2.218F-的捕获 从靶水回收瓶压出18F-通过 QMA柱,18F-被捕获在QMA柱上,这时内置在合成模块中的放化活度探头RI1可以测量出捕获到的18F-放射性活度,记录此数值为回旋加速器的产量值。
1.2.318F-的洗脱和干燥 用 0.9 mL K2.2.2/K2CO3溶液(22 mg K2.2.2溶于0.7 mL乙腈/7 mg K2CO3溶于0.2 mL水)将吸附在QMA柱上的18F-淋洗至反应管,记录此时设备中参数RI2的值,记为参加反应的18F-活度。然后开始加热反应管除去体系里面的水和乙腈。蒸干后继续向反应瓶中加入0.5 mL无水乙腈,再次加热蒸干,因为水的存在会使氟化取代反应的合成效率大幅度降低,通过两次除水,确保反应体系里没有水的存在。
1.2.4加入前体NITTP进行氟化反应 在反应管中加入1.5 mL无水乙腈溶解的10 mg前体1-(2’-硝基-1’-咪唑基)-2-氧-四氢吡喃基-3-氧-甲苯磺酰基-丙二醇(NITTP),溶液添加完毕后通入氮气鼓泡30 s,确保体系混合均匀,在密闭的反应管内120 ℃加热反应,压力传感器监测到反应管内的压力在100~110 kPa。7 min冷却至室温,视频监控中观察反应液的颜色变为淡黄色。
1.2.5水解和中和 向反应液中加入0.75 mL 1.0 mol/L的盐酸溶液,120 ℃下密闭加热反应4 min后冷却至室温,加入1.5 mL Na2HPO4和NaH2PO4组成的缓冲溶液(pH=7~8)进行中和。
1.2.6产品的HPLC纯化 将粗产品进样到半制备型HPLC中进行分离纯化,流动相为V(乙腈)∶V(水)=15%∶85%,用放射性检测器和紫外检测器进行监测,流速3 mL/min,紫外检测波长325 nm,18F-FMISO产品在11 min左右出放射性峰(图2中尖锐峰A),收集此时的产品进入加有稳定剂的旋转蒸发仪中,用1 mL无水乙醇清洗管壁上残留的产品进入旋转蒸发仪的梨形烧瓶,加热并在真空状态下脱除溶剂,80 ℃下加热80 s,再升温至130 ℃下加热直至溶剂完全蒸发,注入10 mL生理盐水稀释溶解,过0.22 μm无菌滤膜,得到产品。合成的关键步骤在于半制备型HPLC对产品的分离,合成前先以流动相冲洗半制备色谱柱,观察紫外检测器和放射性检测器的基线是否平稳、柱压是否稳定、管路连接处是否有漏液现象。合成进行到“HPLC INJECT”步骤,HPLC的泵自动启动,通过摄像头或者铅玻璃观察反应液是否完全被注射进入HPLC的“LOOP”环内,合成软件自动记录此刻为起始时间,当11 min左右出现尖锐放射性峰时开始收集产品,当放射性峰形回落至基线状态,停止收集。
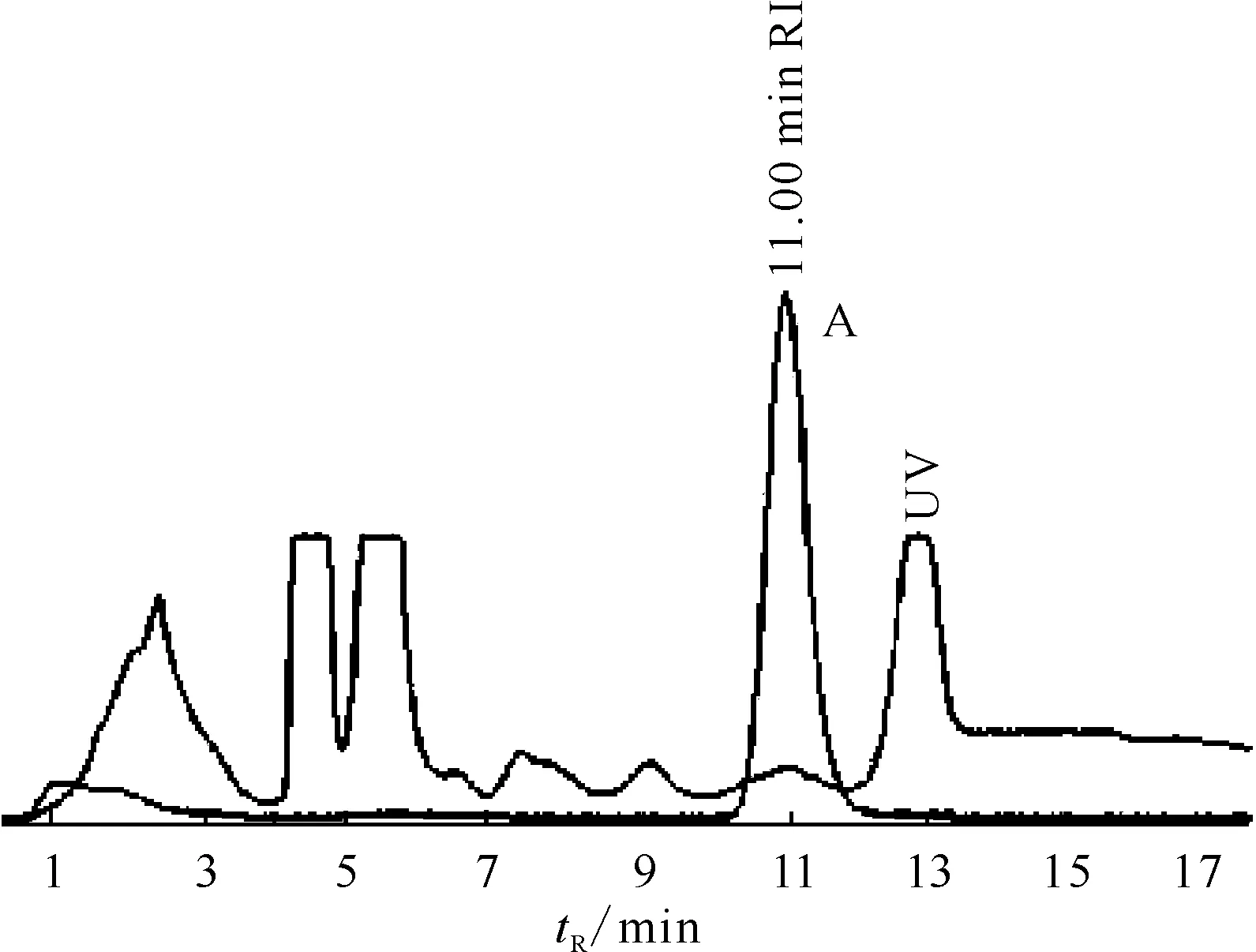
图2 15%乙腈为流动相时18F-FMISO的HPLC分离曲线
1.3 质控
1.3.1薄层色谱法 用毛细玻璃管蘸取微量的产品溶液,在2×10 cm硅胶板上点样,然后放置到展开缸中展开,展开剂为甲醇,用放射性薄层色谱仪检测放化纯度。
1.3.2高效液相色谱法 用5 mL注射用水溶解1 mg19F-FMISO标准品,用微量注射器抽取20 μL,注入到质控型HPLC进样器内,以V(乙腈)∶V(水)=8.5∶1.5做为流动相,流速1 mL/min,紫外检测波长325 nm,柱温35 ℃。用紫外检测器和放射性检测器分别检测产品的化学纯度和放化纯度。
1.3.3氨基聚醚(K2.2.2)的含量检测 参考《中华人民共和国药典》2015年版第二部[5]氟[18F]脱氧葡糖注射液中氨基聚醚(K2.2.2)的检测方法,对产品进行K2.2.2含量的测定。
1.3.4细菌内毒素的检测 参考《中华人民共和国药典》2015年版第四部通则1143[6]105-109进行细菌内毒素检测。
1.3.5溶剂残留检测 参考《中华人民共和国药典》2015年版第四部通则0861[6]154-157残留溶剂测定法,以硝基对苯二酸改性的聚乙二醇为固定液的毛细管柱为色谱柱,柱温70 ℃,进样温度200 ℃,检测器温度为250 ℃,顶空瓶温度为85 ℃,平衡时间10 min,取0.5 mL样品进样进行检测。
1.3.6放化稳定性研究 用HPLC法,采用紫外和放射性双检测器,检测抗坏血酸和抗坏血酸钠做为稳定剂的情况下,产品的放化纯度和时间的关系。记录合成结束后时间为起始时间,每间隔一定时间取样,检测产品的放化纯度。
2 结果与讨论
采用住友CFN MPS-200合成装置,NITTP为前体化合物,与回旋加速器生产出的18F-经氟化取代后水解,加缓冲溶液调节pH值后用半制备型HPLC分离,再进入旋转蒸发仪脱掉溶剂,加生理盐水溶解产品,转出后过无菌滤膜得到产品。整个合成时间50 min,不校正合成效率EOS=(45±5)%(n=20)。透过铅玻璃观察产品为无色或淡黄色透明溶液,放置10个半衰期后取出观察,产品为无色透明溶液。用精密pH试纸测定产品pH值在6~8之间。HPLC法检测,产品和标准瓶溶液的保留时间一致,K2.2.2的质量浓度小于25 mg/L,1 mL的细菌内毒素含量小于15EU,产品中乙醇的质量分数小于1.5×10-3;乙腈质量分数小于15×10-6,未检出丙酮成分,符合临床使用要求。
2.1 制备型HPLC流动相选择
分别选用15%(体积分数,下同)的乙腈(见图2)和10%、5%的乙醇做为流动相进行产品纯化,结果示于图3—5。图3结果表明,以10%的乙醇做为流动相分离产品时,产品的保留时间为9.67 min(如图3中尖锐峰A),但分离度较差,放射性峰与杂质紫外峰重合,用质控型HPLC对产品进行化学纯度检测发现,在保留时间为6.322 min和6.598 min(见图4)时出现两个杂质吸收峰,产品的化学纯度无法满足要求。图5结果表明,以5%的乙醇做为流动相分离产品时,产品的保留时间为20.33 min(如图5中尖锐峰A),虽然分离效果较好,但保留时间过长,影响合成效率。
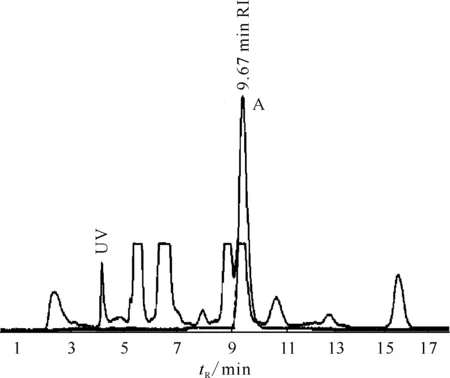
图3 10%乙醇为流动相时18F-FMISO的分离曲线

图4 10%乙醇为流动相进行18F-FMISO分离时得到产品的HPLC检测UV曲线
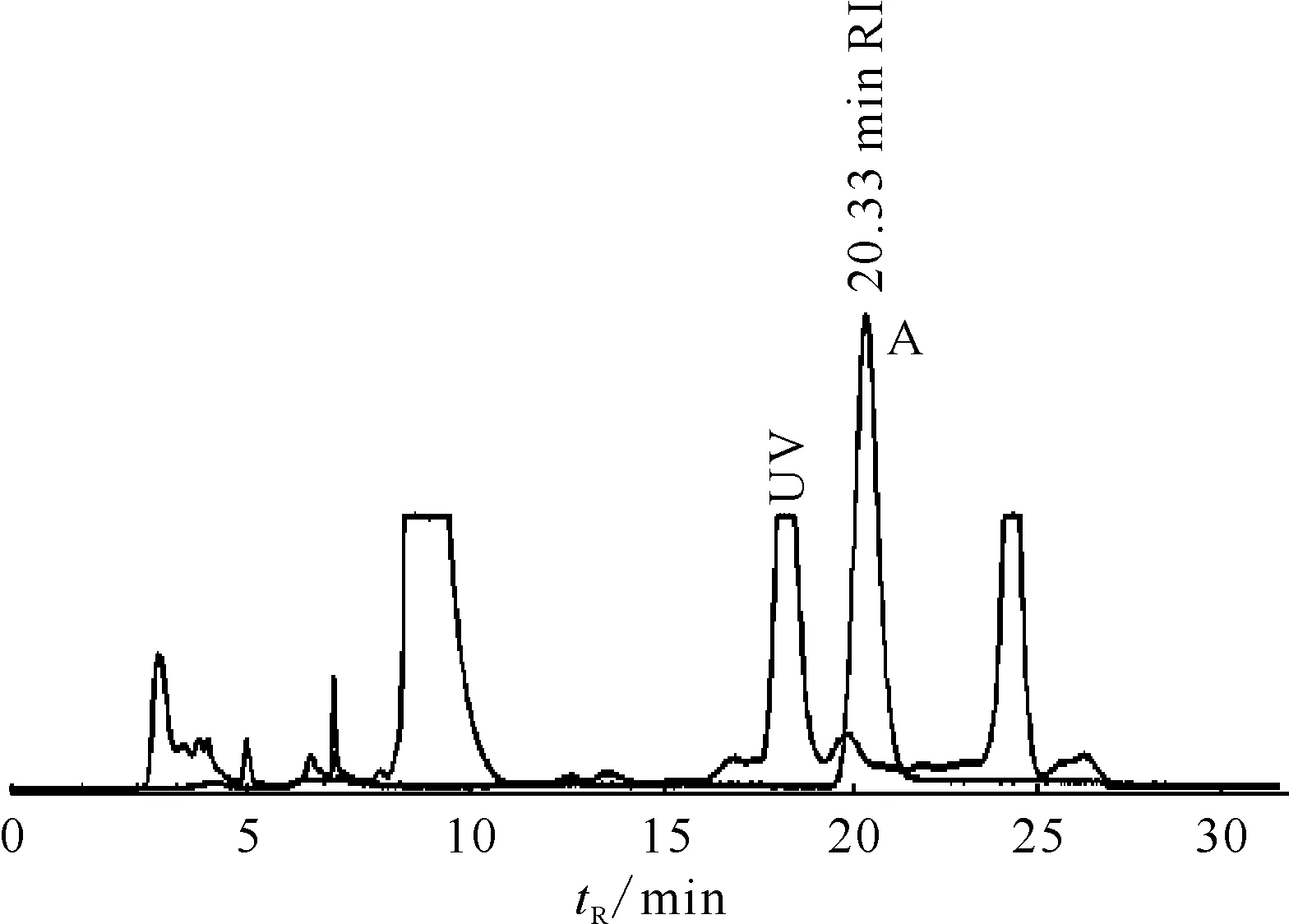
图5 5%乙醇为流动相时18F-FMISO的分离曲线
对比乙醇和15%乙腈水溶液(图2)做流动相进行产物分离的结果表明,15%乙腈作为流动相的分离效果优于乙醇,且重复性好,可以在3 mL/min的低流速下分离产物,柱压低。经旋蒸后除去乙腈溶剂,测得乙腈质量分数小于15×10-6,远低于国家药典对氟代脱氧葡萄糖(FDG)中乙腈质量分数小于4×10-4的要求。因此,在有旋转蒸发仪的条件下,可以选择15%乙腈水溶液作为流动相,为18F-FMISO的纯化增加一种工艺路线选择。以下实验均采用15%乙腈水溶液作为流动相。
2.2 18F-FMISO的放化稳定性
制备型HPLC分离出的产品溶液进入旋转蒸发仪的梨形烧瓶,加热并在负压状态下脱除溶剂,80 ℃加热80 s,再升温至130 ℃加热直至溶剂完全蒸发,注入10 mL生理盐水稀释溶解产品。在不添加任何稳定剂的情况下,测得18F-FMISO的放化纯度与时间的关系,结果列于表1。从表1可以看出,产品无法达到放化纯度大于95%的标准,但辐射自分解的速率较慢,6 h后的放化纯度只降低了2~3个百分点。辐射自分解的原因是高能射线产生的自由基将水分解成自由的质子、羟基和过氧化氢,其中过氧化氢的质量浓度可达2 mg/L,这些质子、羟基和过氧化氢破坏目标产物的分子结构,从而导致产品分解[7]。
迄今为止,辐射自分解的机理并不明确,但可通过18F-FMISO分子结构进行解释。在18F-FMISO分子中,18F-的电负性比碳原子大,18F-吸引电子的结构C—18F键之间的电子云密度偏向于18F-,碳原子带有部分正电荷。因此与18F-相连的碳原子更容易被亲核试剂进攻,由于连接18F的碳原子为伯碳原子,故易于发生SN2亲核取代。
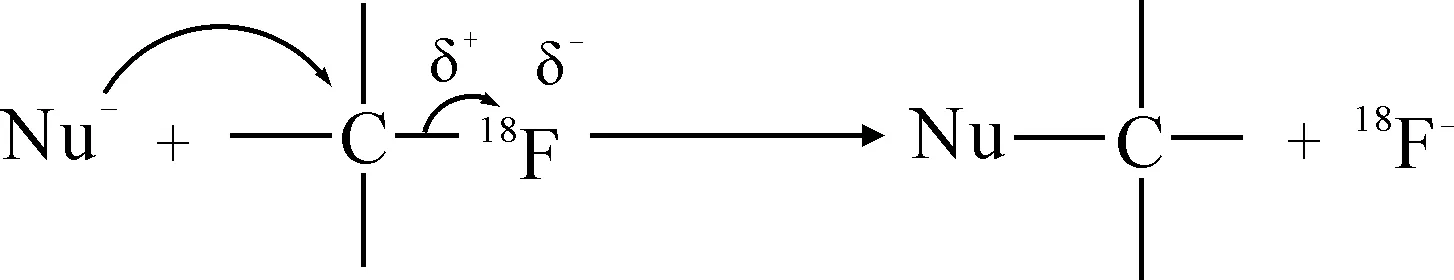
SN2亲核取代反应的特点之一是反应速率与两种反应物的浓度都相关,高活度的产品辐射分解产生更多的自由的质子、羟基和过氧化氢,从而加快18F-FMISO的辐射自分解。从表1可见,随着产品活度的增大,产品经旋蒸处理后放化纯度逐渐变低,活度的大小是影响18F-FMISO放化稳定性的第一因素,活度越大,单位时间内形成的质子、羟基、过氧化氢越多,对C—18F键破坏越严重。
SN2亲核取代为吸收能量反应,当旋蒸加热温度超过C—18F键断裂所需要能量时,加快了18F-FMISO的分解。实验表明,将放化纯度大于99%的18F-FMISO含乙腈的水溶液在80 ℃下加热至溶剂蒸发完全(约用时6 min)后取样进行HPLC分析,放化纯度降低为91.3%。所以,在不加稳定剂的条件下,直接采用旋转蒸发仪脱除分离产品溶液中的乙腈溶剂,会导致产品脱氟而放化纯度无法达到使用要求。研究了较低温度(50 ℃)下,不添加任何稳定剂,利用旋转蒸发仪在高真空的作用下蒸发溶剂,300 s后转出产品,立刻检测放化纯度,发现产品的放化纯度依然小于95%,由此可以判断,温度是影响产品放化纯度降低的重要因素。
2.3 18F-FMISO放化稳定剂的选择
选择抗坏血酸做稳定剂,在旋转蒸发仪的梨形烧瓶中预先加入0.1 mL 100 g/L的抗坏血酸水溶液和1 mL无水乙醇。在HPLC分离出产品溶液进入旋转蒸发仪的梨形烧瓶内,与抗坏血酸溶液和乙醇均匀混合后开始加热,负压下蒸发溶剂,80 ℃加热80 s,再升温至130 ℃加热直至溶剂完全蒸发,加入10 mL生理盐水溶解,得到产品后定时取样进行放化纯度分析,结果列于表2。

表2 添加抗坏血酸稳定剂条件下18F-FMISO放化纯度与时间的关系
从表2可以看出,在抗坏血酸和乙醇共同存在下,加热18F-FMISO的乙腈水溶液,稳定剂不仅没有起到作用,相反产品的稳定性更加恶化,辐射自分解情况非常严重。抗坏血酸不适宜作为此合成工艺下的稳定剂,这与文献[8]中抗坏血酸做为稳定剂的情形不符。表2中三批次产品的放化纯度相差很大,这可能是由于旋蒸时加热温度不均,或操作者对干燥程度判断差异引起的加热时间长短不同所致。
Scott等[9]认为,正电子湮灭产生的高能射线引起水性介质产生了羟基自由基和活性氧,正电子示踪剂的分解源于两种物质的作用。并提出稀释产品降低比活度的方法可以减缓正电子示踪剂的分解速率而无法阻止其分解。他研究了抗坏血酸钠做为稳定剂的优势为对产品溶液的pH影响较小,缺点为用碘铂酸钾TLC点样法,对产品进行K2.2.2含量检测时,可能出现假阴性。
本工作选择抗坏血酸钠做为稳定剂,加入量与加入方法与抗坏血酸做稳定剂的实验条件相同,结果列于表3。从表3可以看出,抗坏血酸钠的加入,可以保护产品的放化稳定性,130 ℃加热至溶剂挥发完全后放化纯度大于98%,在6 h后放化纯度仍然大于95%。

表3 添加抗坏血酸钠稳定剂条件下18F-FMISO放化纯度与时间的关系
联合使用抗坏血酸和抗坏血酸钠做为稳定剂,在旋转蒸发仪的梨形烧瓶中预先加入0.05 mL 100 g/L的抗坏血酸水溶液、0.05 mL 100 g/L的抗坏血酸钠水溶液和1 mL无水乙醇。负压下蒸发溶剂,80 ℃加热80 s,再升温至130 ℃加热直至溶剂完全蒸发,得到产品后定时取样进行HPLC分析,测得放化纯度为80.5%。
表4为pH值对18F-FMISO放化稳定性的影响。由表4可见,pH值是影响18F-FMISO放化稳定性的另一因素,18F-FMISO在中性及弱碱性体系内稳定,而在酸性体系内稳定性差。

表4 pH值对18F-FMISO放化稳定性的影响
2.4 18F-FMISO的纯度检测
用5 mL注射用水溶解1 mg标准品19F-FMISO,以微量进样器吸取20 μL进行HPLC检测,结果示于图6。由图6可知,标准品19F-FMISO的保留时间为6.097 min。在相同的条件下进行18F-FMISO的HPLC放射性检测,结果示于图7。如图7所示,18F-FMISO的保留时间为6.109 min,F-的保留时间为2.848 min,产品18F-FMISO的保留时间和标准品19F-FMISO的保留时间基本一致。图8为HPLC法检测18F-FMISO的化学纯度曲线。由图8可知,保留时间为2.743 min的紫外吸收峰为抗坏血酸钠;6.115 min为产品18F-FMISO的吸收峰,但成品溶液中18F-FMISO的化学含量非常低。

图6 HPLC法检测19F-FMISO标准品的UV曲线
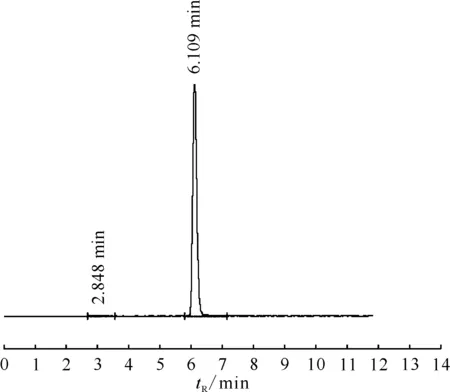
图7 18F-FMISO的放化纯度检测曲线

图8 HPLC法检测18F-FMISO的化学纯度曲线
用薄层色谱法检测产品的放化纯度,结果示于图9。F-的放射性峰在原点,产品18F-FMISO的Rf=0.575 min,放化纯度大于95%。

图9 薄层色谱法检测18F-FMISO的放化纯度
3 结 论
(1)回旋加速器生产出的18F-经QMA柱捕获后由K2CO3和K2.2.2的混合溶液淋洗,经两次干燥除水后加入乙腈溶解的前体NITTP,在完全密闭加热下进行氟化取代反应,冷却后加盐酸水解去除保护基团,再以缓冲溶液中和反应体系,用半制备HPLC以V(乙腈)∶V(水)=15%∶85%为流动相进行产品分离,将产品溶液转入预先加入抗坏血酸钠的梨形烧瓶内,加热脱除溶剂,最后加入生理盐水溶解,过0.22 μm无菌滤膜得到18F-FMISO产品。引起18F-FMISO分解有三个主要因素:活度、温度和pH。18F-FMISO稳定性与活度、温度成反相关;在中性及弱碱性溶液中稳定,在弱酸性溶液中容易分解。
(2)采用抗坏血酸钠做为稳定剂,80 ℃加热80 s后升温至130 ℃下加热至溶剂完全蒸发,产品能保持良好的放化稳定性,6 h后放化纯度依然大于95%。通过旋蒸处理,产品中只含有微量的乙醇、乙腈溶剂,毒副作用小。合成时间50 min,不校正合成效率EOS=(45±5)%(n=20),整个过程由合成模块全自动合成,无需人为干预,满足18F-FMISO的实验和临床应用需求。
