以皮肤骨瘤为首发症状的Albright遗传性骨营养不良症一例
2020-10-24寒宇阳赵文英李垣君张杏莲
寒宇阳 赵文英 李垣君 张杏莲
1山西省儿童医院皮肤科,太原030013;2山西省儿童医院病理科,太原030013
患儿女,4月龄,因左腹部、大腿后侧暗红色斑块2个月就诊。患儿出生后2个月,父母发现其左腹部、大腿后侧有暗红色斑块,未发现患儿有不适症状。患儿系足月顺产,纯母乳喂养,无宫内发育迟缓,无窒息史,无其他疾病史。父母均体健,母亲身高149 cm,父亲身高168 cm,均无类似疾病史,非近亲结婚。家族中无类似疾病患者。
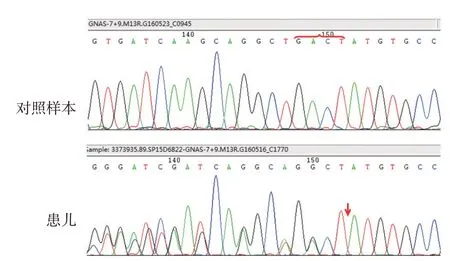
体检:一般情况良好。身高60 cm(4月龄患儿参考值:61.0 ~65.4 cm),体重10 kg(4月龄患儿参考值:6.11 ~7.56 kg),出生时身高体重不详。满月脸,四肢短小,指趾短(图1A)。肺部听诊有轻微喉喘鸣。甲状腺未触及肿大。心脏、腹部未发现明显异常。无癫痫、肢体抽搐、意识丧失等神经系统异常。皮肤科检查:左侧腹部、大腿后侧见4 cm × 3 cm、4 cm × 2 cm 大小不规则、边界不清的暗红色斑块(图1B),可触及皮下颗粒状小结节,骨样硬度,无触痛、红肿,无白色物质排出。实验室检查:血钙2.36 mmol/L、血磷2.36 mmol/L(参考值1.45 ~2.10 mmol/L,下同);甲状旁腺素24.11 pmol/L(1.6 ~6.9 pmol/L)、游离三碘甲状腺原氨酸5.57 pmol/L(5.1 ~8.0 pmol/L)、游离甲状腺素9.92 pmol/L(12.1 ~18.6 pmol/L)、促甲状腺素19.195 mIU/L(0.64 ~6.27 mIU/L)、甲状腺素77.2 nmol/L(77.8 ~170.0 nmol/L)、三碘甲状腺原氨酸2.26 nmol/L(1.8 ~3.68 nmol/L);促肾上腺皮质激素717 ng/L(0 ~46 ng/L),皮质醇274 mmol/L(138 ~690 mmol/L);血尿粪常规、肝肾功能均无异常。腹部皮损组织病理:真皮、皮下组织可见蓝染无结构钙质沉积,周围粉染骨样基质内卵圆形胞质透亮的骨细胞,表皮未见明显异常(图2)。充分告知患儿家长并建议进行基因检查,家长同意并签署知情同意书。抽取患儿外周血送至太原金域临床检验有限公司基因检测,结果显示,GNAS 基因第7 外显子上568 ~571 位GACT碱基缺失(c.568_571delGACT)杂合突变(图3)。
诊断:Albright遗传性骨营养不良症(Albright hereditary osteodystrothy,AHO)。
治疗:家长拒绝治疗,电话随访,至今无明显变化。

讨论AHO 的临床特征包括皮肤和皮下组织异位骨化、满月脸、肥胖、矮小、短指趾畸形(特别是第4和第5掌指骨畸形),伴内分泌系统紊乱包括假性甲状旁腺功能减退(pseudohypoparathyroidism,PHP)[1⁃2]。AHO 患者甲状旁腺素分泌正常但机体对甲状旁腺素应答有缺陷时表现为PHP[3],临床常以反复发作的手足抽搐、感觉异常、癫痫样发作等为首发症状,血清学检查表现为低血钙、高血磷及血甲状旁腺素升高。有学者认为[4⁃5],皮肤骨瘤是AHO的重要体征,即使患者的血清生化值正常也不能排除AHO 的可能性。AHO的病理组织学表现为真皮或皮下结缔组织钙化及新生骨形成且骨化不进行性发展,头颅CT有时可见基底节及灰、白质交界区多发钙化。本例患儿以皮肤骨瘤为首发症状,具备AHO 典型的临床体征,病理检查可见钙质沉积及骨样组织形成,实验室检查不仅有甲状旁腺素升高,还有促肾上腺皮质激素和促甲状腺激素升高,分子遗传学检查也支持AHO诊断,且本患儿随访至今并未出现进行性发展的症状,因此AHO诊断明确。患儿父母拒绝做脑部核磁检查,故未能观察患儿脑部是否有钙化情况。
GNAS 基因主要产物是编码调控腺苷酸环化酶活性的G 蛋白激活因子α 亚基(Gsα)[6],其移码突变可使其Gsα 活动度降低,导致蛋白功能丧失[7],造成患者甲状旁腺素及其他内分泌激素抵抗。AHO 的致病突变以GNAS 基因第7 外显子上4 个碱基对的缺失常见,其他突变大多为个体特异性[8]。本例患儿基因检测结果显示,GNAS 基因第7 外显子c.568_571delGACT杂合突变,属于常见突变位点,与国外报道的突变位点一致[9]。GNAS 基因纯合突变在人类中还未曾有过报告,可能因为纯合突变为致死性[10]。GNAS基因具有基因组亲源印迹特点[11⁃12],突变基因来自母亲可导致AHO[13]。本例患儿父亲身高正常,母亲身高低于正常成年女性,因此怀疑该患儿突变基因可能来源于其母亲,但由于经济原因,父母不同意进行相关实验室检查及基因检测。
本病在临床上需与进行性骨化性纤维发育不良、进行性骨发育异常、板样皮肤骨瘤等疾病鉴别。进行性骨化性纤维发育不良结缔组织的骨化为进行性且无内分泌异常,其皮肤和皮下组织受累常由深部病变扩大蔓延所致,异位骨化发生前有典型的炎症反应,与ACVR1 基因突变有关。进行性骨发育异常的皮损常在生后不久出现,可快速进展,出现深部结缔组织、肌肉的异位骨化,一般无发育或内分泌的异常。板样皮肤骨瘤仅有局限性异位骨化,无其他异常。
关于AHO 的治疗,如骨化已经形成,手术切除新生骨仍是目前主要的治疗方法,若伴有内分泌异常则主要针对潜在的钙磷代谢异常和激素抵抗治疗。本例患儿监护人拒绝治疗,目前电话随访中。
利益冲突所有作者均声明不存在利益冲突
