固相萃取-HPLC柱后氧化衍生荧光法测定运动营养食品中叶酸的含量
2020-10-23黄树楷许伟沂彭丽诗
陈 琼,黄树楷,黄 盼,许伟沂,龙 梓,彭丽诗
(仙乐健康科技股份有限公司,广东汕头 515041)
叶酸(folic acid,FA)即维生素B11,是蝶呤衍生物,也称作蝶呤酰谷氨酸(pteroylglutamic acid,PGA),它可为核苷酸生物合成、氨基酸代谢和脱氧核糖核酸(DNA)甲基化提供单碳基团,能促进骨髓中造血幼细胞成熟,是所有生物机体细胞繁殖和胎儿的发育生长必需的维生素,故叶酸的缺乏可导致心血管疾病、神经系统疾病、消化系统疾病、诱发癌症等,如巨幼红细胞贫血、白血球减少症、新生儿神经管畸形、智力退化、免疫力降低等[1-7]。因此,在许多的食品中,尤其是营养素补充剂和运动营养食品都把叶酸作为重要的添加物质。
通常运动营养食品中,叶酸的添加量低,基质较为复杂,常见配方中不仅含有牛奶、乳清及大豆等高蛋白的配料,而且还附带谷物、水果等干扰成分,因此样品前处理难度较大。而目前测定叶酸常用的方法有:比色法[8-10]、薄层层析法[11]、微生物法[12-16]和色谱分析法[17-23]等,其中比色法、薄层层析法虽操作相对简单,但对于复杂基质样品而言易受干扰,导致结果准确度不高。微生物法是经典方法,GB5009.211-2014 《食品中叶酸的测定》和EN 14131-2013 《Foodstuffs Determination of folate by microbiological assay》采用微生物法进行测定,灵敏度高,但也存在着许多的局限性,如实验周期较长,方法重复性差,受样品中抑菌成分的影响等[24]。AOAC-2011.06测定婴儿配方奶粉和成人营养素中的总叶酸采用LC-MS/MS法,但由于液质法存在基质效应,会影响其稳定性和结果重现性[25-26],且液质相对检测时间长,操作较为繁琐。液相色谱法实现了叶酸与其他成分的分离,特异性高,但通常运动营养食品中叶酸的添加量较其他水溶性维生素低,若使用紫外检测器时须加大取样量,从而造成较为严重的干扰[27]。综上,开发出操作便捷、灵敏度高、方法重复性及专属性好的运动营养食品叶酸测定方法,有着重要的意义。
本文在现有的检测方法基础上,考察了沉淀蛋白法、酶解蛋白法、固相萃取法三种样品前处理方法对比,并优化了提取溶剂与流动相的条件,以最优条件对运动营养食品中叶酸进行富集和纯化,再让弱荧光效应的叶酸经色谱柱分离,利用柱后衍生仪使叶酸与强氧化剂过二硫酸钾进行反应生成有强荧光性的蝶呤-6-羧酸(如图1),最后通过荧光检测器检测,外标曲线法定量。
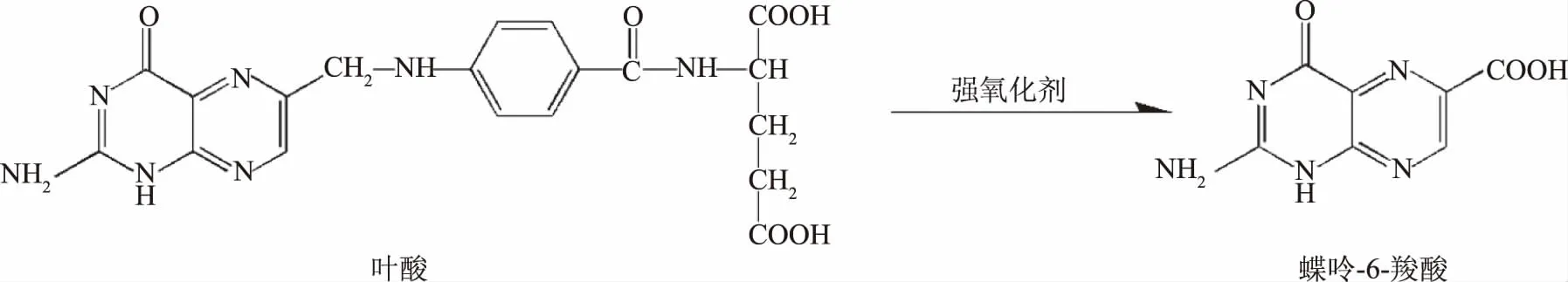
图1 叶酸的氧化反应方程式
1 材料与方法
1.1 材料与仪器
运动能量棒(标签叶酸含量19 μg/g)、运动代餐粉(标签叶酸含量15 μg/g)、运动代餐奶昔(标签叶酸含量21 μg/g) 天猫旗舰店;反相弱阴离子交换Strata-X-AW固相萃取小柱(60 mg/3 mL) 美国Phenomenex公司;反相硅胶吸附C18固相萃取小柱(20 cc)、混合模式阴离子交换 Oasis MAX固相萃取小柱(20 cc) 美国Waters公司;叶酸标准品 含量95.3%,中国食品药品检定研究院;乙腈 色谱纯,美国TEDIA公司;磷酸二氢钾、磷酸、氨水 色谱纯,德国CNW公司;盐酸 分析纯,西陇科学;甲醇 分析纯,广州化学试剂厂。
1260型高效液相色谱仪(配荧光检测器) 美国Agilent公司;Pinnacle PCX柱后衍生仪 美国Pickering公司;Tissuelyser-24冷冻研磨 上海净信实业发展有限公司;CASCADA I型超纯水机 美国PALL公司;XS205DU型分析天平 瑞士METTLER-TOLEDO公司;UNIVERSAL 320R型离心机 德国Hettich公司;DZKW-D-2型水浴锅 上海科恒实业发展有限公司。
1.2 实验方法
1.2.1 试剂的配制 提取剂:取2.5 mL浓氨水,加水约950 mL,用盐酸调节pH至8.5,加水定容至1000 mL。
50 mmol磷酸二氢钾(pH3.5):取磷酸二氢钾6.8 g溶于950 mL水中,用磷酸调节pH至3.5,加水定容至1000 mL。
5%过二硫酸钾溶液:取5 g过二硫酸钾,加水定容至1000 mL。
1.2.2 标准溶液配制及标曲绘制 精密称取50 mg叶酸标准品至50 mL容量瓶中,用提取剂溶解并稀释得到浓度为1 mg/mL的标准储备液。分别吸取标准储备液用水稀释得到浓度分别为0.2、0.4、0.8、1.2、1.6、2.0 μg/mL的标准曲线溶液。分别吸取标准溶液和样品溶液适量,按色谱条件进样分析,根据保留时间定性,标准曲线法定量。
1.2.3 样品前处理方法的选择
1.2.3.1 沉淀蛋白法 准确称取粉碎均匀的试样于烧杯中,用提取剂溶液60 ℃溶解后,转入容量瓶中。加入10 mL 13%高氯酸溶液,混合均匀后静止20 min。用提取剂定容至刻度,混匀离心,上清液过0.45 μm微孔滤膜后上机测定。
1.2.3.2 酶解蛋白法 准确称取粉碎均匀的试样于离心管中,加入适量蛋白酶与α-淀粉酶,提取溶剂适量,37 ℃水浴振摇2 h,转入容量瓶中,用提取剂定容至刻度,混匀过滤,滤液过0.45 μm微孔滤膜后上机测定。
1.2.3.3 流动相优化 选用50 mmol/L磷酸二氢钾水溶液∶乙腈=90∶10 (V/V)为流动相,保持流动相比例不变,用磷酸分别调50 mmol/L磷酸二氢钾水溶液的pH为2.0、2.5、3.0、3.5、4.0、4.5、5.0、5.5、6.0、6.5、7.0、7.5、8.0、8.5,进样测定。
1.2.4 样品溶液的制备 由于运动营养食品中通常含有谷物粒、坚果碎、水果粒等颗粒较大的成分,且样品本身黏度较大,若直接用粉碎机粉碎,会造成样品粉碎不均匀且易黏附粉碎刀头,造成卡机。本试验采取液氮冷冻的方式对样品进行预处理,再用冷冻球型研磨仪进行粉碎,保证了样品粉碎的均匀性。样品前处理具体操作如下:精密称取经液氮冷冻粉碎均匀的样品5~10 g至50 mL烧杯中,加入约25 mL提取剂,60 ℃水浴20 min,溶解后冷却至室温,用氨水或盐酸调节pH至8.0±0.5,超声提取10 min,时时振摇,转移到50 mL容量瓶,用提取剂定容至刻度,5000 r/min离心10 min,精密吸取样品上清液溶液1 mL,上样于反相弱阴离子交换Strata-X-AW固相萃取小柱(依次用2 mL甲醇和2 mL水活化),待自然流干后,依次加入1 mL的25 mmol乙酸铵溶液(pH6~7)、1 mL甲醇淋洗柱床,弃去洗脱液。再每次用1 mL 5% NH4OH甲醇溶液进行洗脱2次,待自然流干后,吹出柱床内所有溶液并收集洗脱液,60 ℃以下氮吹至干,加提取剂定容至1 mL,摇匀,过0.45 μm微孔滤膜,待测。整个实验过程应注意避光操作。
1.2.5 色谱条件 检测器:荧光检测器;检测波长:激发波长365 nm,发射波长440 nm;柱前流动相:50 mmol磷酸二氢钾(pH3.5)∶乙腈=90∶10;流速:1.0 mL/min;色谱柱:Venusil MP C18,4.6 mm×250 mm,5.0 μm;柱温:30 ℃;进样量:10 μL;柱后衍生剂:5%过二硫酸钾溶液;衍生体积:0.5 mL;衍生温度:60 ℃;衍生仪流速:0.3 mL/min。
1.3 数据处理
色谱结果积分处理采用1260型高效液相色谱仪配套的Agilent ChemStation色谱工作站,并利用Excel软件进行数据统计及数据分析。
2 结果与分析
2.1 样品前处理优化
2.1.1 沉淀蛋白法 高蛋白含量样品的前处理中,沉淀蛋白并提取目标物是最常用手段,而效果较佳常见的蛋白沉淀剂有亚铁氰化钾和乙酸锌、三氯乙酸和高氯酸等。亚铁氰化钾和乙酸锌混合会生成亚铁氰化锌,可与蛋白质共沉淀,但同时也会与少部分叶酸结合沉淀,造成目标物的损失。而三氯乙酸则会破坏叶酸的蝶啶环,使后续氧化衍生无法进行,高氯酸既可兼顾除蛋白质,又可保证叶酸的蝶啶环不被破坏,故选择高氯酸沉淀剂进行实验。3组平行实验测定结果如表1,实验发现沉淀蛋白法回收率在78.7%~88.4%之间,去除蛋白效果虽好,但仍有部分目标物损失,可能原因是在低于蛋白质等电点的pH下,蛋白质阳离子与高氯酸阴离子下形成难溶性盐,蛋白变性从而沉淀,该沉淀形成过程迅速,导致沉淀物内部包裹住少量叶酸分子,形成包埋,导致部分叶酸损失。
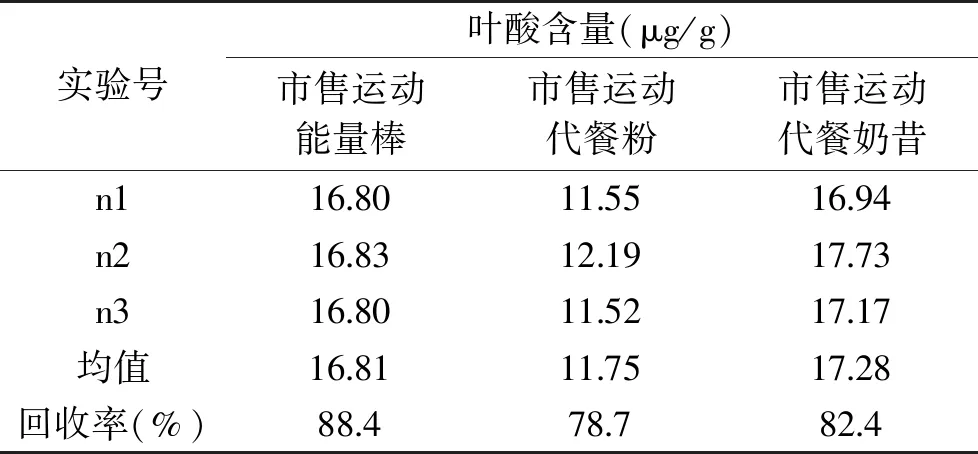
表1 沉淀蛋白法测定叶酸含量
2.1.2 酶解蛋白法 叶酸标准品色谱图见图2,酶解样品色谱图见图3,结果发现蛋白被酶解成多肽、氨基酸等亦进入样品溶液,使目标峰被干扰,酶解法前处理效果不理想。可能的原因为蛋白酶是酶解蛋白最常见的酶之一,其作用机理是识别肽键两侧的氨基酸残基的特定R基,与之结合使肽键断裂[28],从而使蛋白大分子瓦解。而淀粉酶是重要的淀粉水解酶,其作用于淀粉时从淀粉分子的内部切开α-1,4糖苷键,生成糊精和还原糖[29-30],水解下来的小分子在该激发波长与发射波长下同样有荧光吸收,从而造成了目标峰被干扰。
图2 叶酸标准品色谱图
图3 酶解法样品色谱图
2.1.3 固相萃取法(C18、MAX、STRATA-X-AW) 固相萃取能有效地将分析物与干扰组分分离,减少目标色谱峰的干扰,是分离净化常用手段。选用市售C18小柱、MAX小柱、STRATA-X-AW小柱对运动能量棒样品溶液进行SPE纯化,结果以目标物的加标回收率作为比较依据。结果发现,相比其他两款小柱,反相弱阴离子STRATA-X-AW小柱对样品提取液的净化效果最好,加标回收率最高,主要是因为在弱酸性条件下,STRATA-X-AW小柱能够吸附具有一定酸性的叶酸目标物,淋洗杂质后,再进行洗脱,洗脱液较为干净,杂质少,起到很好的净化效果。
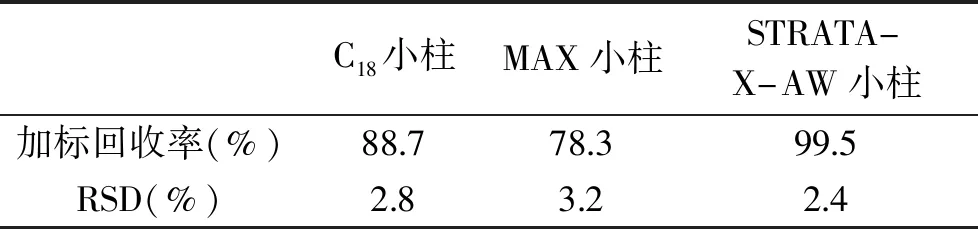
表2 不同固相萃取柱回收率
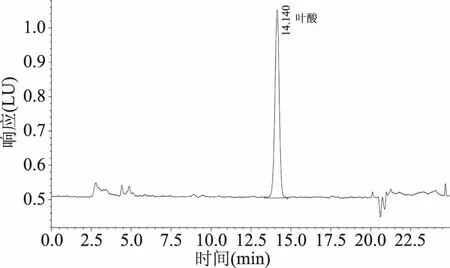
图4 STRATA-X-AW小柱净化样品色谱图
2.2 流动相优化
叶酸水溶性较差,水中溶解度只有1.6 μg/mL,常用0.5%氨水溶液(pH约10)作为提取剂使叶酸转化成叶酸盐,增加其溶解度及稳定性。实验中发现流动相磷酸盐缓冲液的pH对衍生反应产物保留能力及稳定性有一定的关联,分离结果表明,流动相pH在2.0~3.5时,pH越小叶酸的保留时间越往后延,且此时体系缓冲能力不够,从而导致重复性不佳。当pH在3.5以上对叶酸的保留时间影响较小,重复性仍然不佳。但是pH越大,基线越不平稳。而低pH对色谱柱、色谱系统的寿命也有影响。当pH=3.5时,体系缓冲能力刚好能符合重复性要求。综合考虑,实验使用盐酸调节其提取剂pH为8.5,同时选用pH为3.5的磷酸二氢钾水溶液∶乙腈=90∶10 (V/V)作为流动相,既能使得目标溶液处于缓冲液能力范围内,保留时间稳定,又能使得基线稳定,对色谱系统无影响,能满足检测色谱条件要求。
2.3 方法验证
2.3.1 标准曲线、线性范围与仪器定量限 在最佳色谱条件下,检测一系列叶酸标准溶液(0.2~2.0 μg/mL),以标准溶液的浓度为横坐标,以峰面积为纵坐标,绘制线性回归标准曲线,结果发现方法在0.232~2.318 μg/mL范围内呈良好的线性关系,回归方程为y=12.496x-0.04687,决定系数R2=0.9971。以10倍信噪比(S/N)计算,进样量为10 μL,该方法的仪器定量限为0.0774 μg/mL。
2.3.2 重复性、专属性及回收率 按最佳色谱条件,对市售运动能量棒样品进行6 次检测。结果表明该方法重现性较好,峰面积相对标准偏差为2.5%,测定结果较为稳定。分别取阴性样品各9份,每份5 g,分成3组,每组平行3个样,加入不同体积的标准储备液,分别配制成与待测样品中叶酸80%、100%、120%含量的回收率样品,按照方法测定叶酸含量,测定结果如表3,回收率在96.98%~100.50%之间,相对标准偏差在0.98%~3.76%之间,方法回收率良好,准确度较高,阴性样品未检出,见图5,适用于不同基质叶酸的测定。
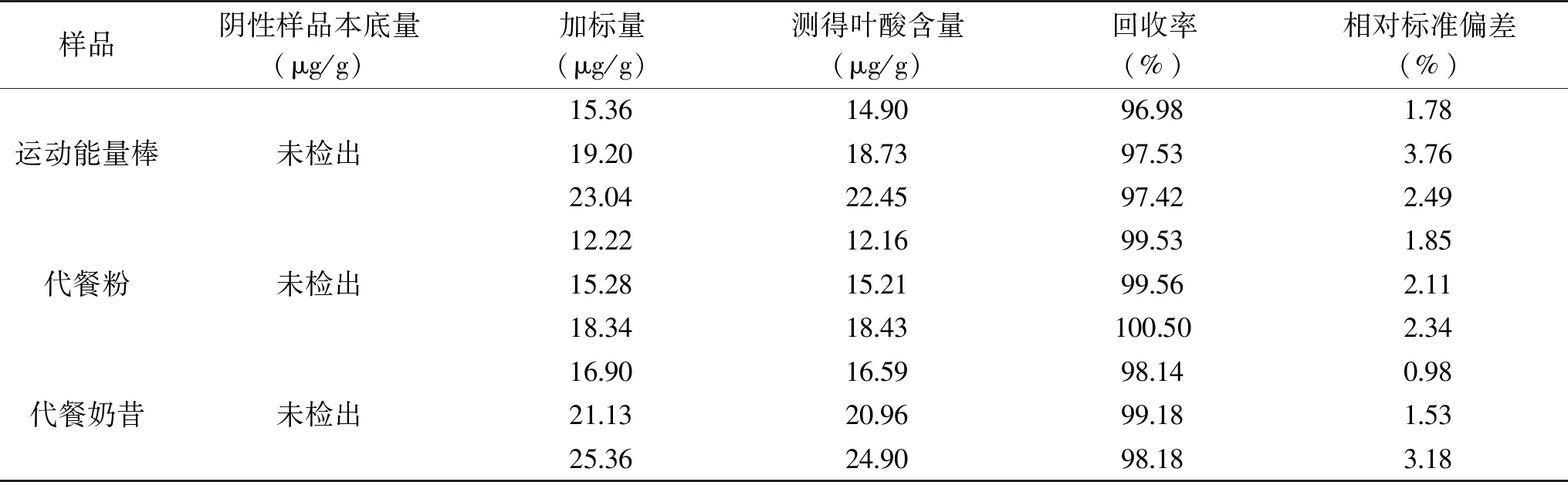
表3 加标回收率测定结果
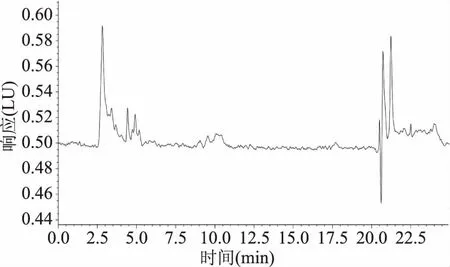
图5 阴性样品色谱图
2.3.3 溶液稳定性 每隔适当时间测定标准品溶液(浓度为1.2 μg/mL)叶酸浓度的百分比及市售样品叶酸含量的百分比,考察溶液稳定性。结果如表4,在该实验条件下,标准品溶解和三款运动营养产品能在8 h内保持稳定(98.54%~102.90%),可满足检测要求。
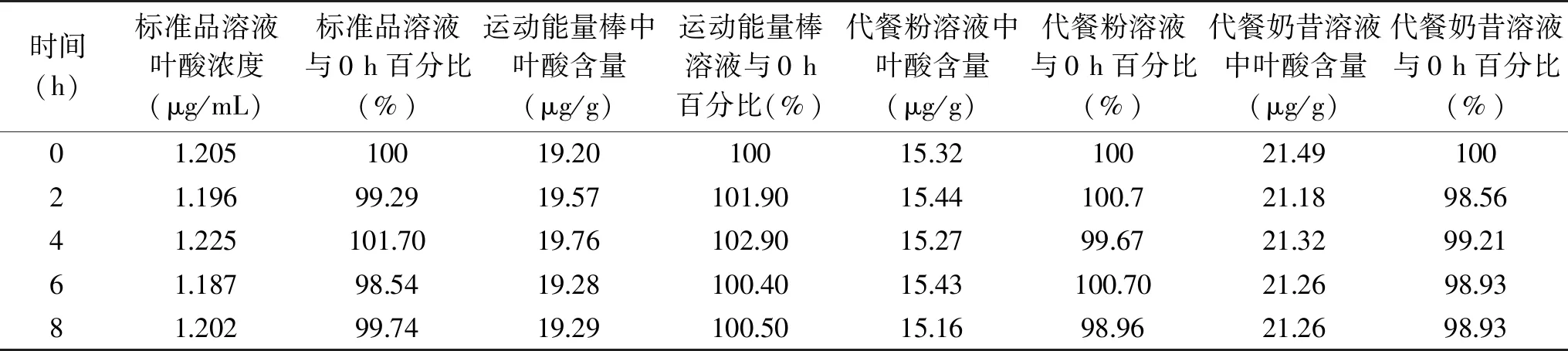
表4 标准溶液及样品溶液稳定性
3 结论
本文开发了固相萃取-HPLC柱后氧化衍生荧光法测定运动营养食品中叶酸含量的方法。分别考察了沉淀蛋白法、酶解蛋白法及固相萃取法的样品前处理方式及流动相的优化条件,样品经反相弱阴离子固相萃取小柱纯化,C18色谱柱分离,柱后衍生仪氧化衍生后,再经荧光检测器,外标法定量。结果表明,在0.232~2.318 μg/mL范围内方法线性良好,R2=0.9971,进样量为10 μL,方法的仪器定量限为0.0774 μg/mL,灵敏度足以满足运动营养食品中叶酸的检测需要,方法回收率在96.98%~100.50%之间,标准品、样品溶液在8 h内稳定。本方法很好地解决复杂基质的运动营养食品中叶酸的提取、净化除杂等问题。操作方便快捷,结果准确,线性范围广,灵敏度高,重现性、专属性、回收率及稳定性较为满意,能够满足运动营养食品中叶酸含量检测的需求。
