新型苯酚类茂金属配合物的合成及表征
2020-10-23李昊坤韩书亮宋文波
李昊坤,韩书亮,金 钊,宋文波
(中国石化 北京化工研究院,北京 100013)
聚烯烃是应用非常广泛的一种树脂产品,一直以来都是非常重要的高分子合成材料,聚烯烃的发展代表了一个国家石化工业的发展水平,是关系到国计民生的重大支柱产业之一[1-2]。聚烯烃的发展与催化剂的发展息息相关,茂金属催化剂就是其中重要的一种[3-4]。茂金属催化剂是继Ziegler-Natta催化剂之后的一类新型的烯烃聚合催化剂。与传统的Ziegler-Natta催化剂相比,茂金属催化剂结构类型丰富,有着更加精确的调控能力[5-8]。茂金属催化剂以卓越的催化性能,突破传统催化剂的局限,合成出新的高性能产品,引起越来越多的重视[9]。尤其是近年来高端化工材料(如聚烯烃弹性体)亟待发展[1,10],对于新型高性能的茂金属催化剂的需求越来越高,科研人员持续探索和开发新型茂金属催化剂,并研究配体结构对催化活性和选择性的影响,以期获得高性能的新型聚合物[11-14]。
本工作设计合成了一系列具有不同取代基的苯酚类茂金属配合物,通过1H NMR,13C NMR,高分辨质谱(HRMS)谱图及单晶X射线衍射对其结构进行了表征,并对合成过程进行优化,得到了带有不同配体的配合物,为开发出新型高性能的茂金属催化剂奠定基础。
1 实验部分
1.1 原料和试剂
乙酸乙酯、石油醚、盐酸、正己烷、三乙胺、氯化铵:分析纯,北京化工厂;氯甲基甲醚(MOMCl)、超干四氢呋喃(99.9%(w))、氢化钠(60%(w)、分散在矿物油中)、氢化钾(30%(w)~35%(w)、分散在矿物油中)、己炔(98%(w))、苯乙炔(98%(w))、碘化亚铜(98%(w))、双三苯基膦二氯化钯(98%(w))、异丙醇(99.5%(w))、五甲基环戊二烯基三氯化钛(98%(w))、二氯甲烷(99.9%(w)):北京百灵威试剂公司;三异丁基铝(TIBA):1 mol/L己烷溶液,上海麦克林生化科技有限公司。所有金属有机反应都在手套箱中进行,溶剂使用超干溶剂。
1.2 分析和测试
核磁共振数据于Bruker公司AVANCE III HD 400M型核磁共振波谱仪上以氘代氯仿为溶剂在25℃下进行测试;高分辨质谱于Bruker公司ESI-Q/TOF MS型质谱仪上以乙腈分散溶剂测定;晶体结构在Bruker公司Bruker-axs CCD型X射线衍射仪上测定。
1.3 配体的合成
配体的合成利用Sonogashira偶联反应[15-16]。将2,6-二碘对叔丁基苯酚(5.0 mmol)溶于四氢呋喃(50 mL)中,在冰浴冷却搅拌下加入氢化钠(7.5 mmol),搅拌1 h后,加入氯甲基甲醚(10 mmol),然后升至室温搅拌2 h,待反应完全后加入饱和氯化铵溶液淬灭,并用乙酸乙酯萃取,减压除去溶剂后以当量的产率得到甲氧甲基保护的苯酚。将得到的产物溶于三乙胺(50 mL)中,氮气置换后,先后加入不同取代基的乙炔化合物(2.1当量)、碘化亚铜(0.2当量)和双三苯基膦二氯化钯(0.1当量),然后室温搅拌过夜。待偶联完成后,用硅藻土过滤除去不溶性固体杂质并用乙酸乙酯洗涤。滤液旋干后粗产品用柱色谱纯化,得到偶联产物。将偶联产物溶于四氢呋喃/异丙醇的混合溶剂中,加入盐酸(6 mol/L),室温搅拌5 h后,用乙酸乙酯萃取并旋干,得到不同的配体化合物。配体合成过程中,所用己炔、苯乙炔、1-乙炔基萘[17]、2-乙炔基萘[17]、9-乙炔基蒽[18]分别参与偶联反应,制得配体L1~L5,配体合成路线见图1。
1.4 茂金属配合物的合成
配合物的合成于手套箱中进行。将制备的配体化合物溶于二氯甲烷中,室温加入纯氢化钾固体(20当量)后反应3 h。待拔氢反应完全之后,加入五甲基环戊二烯基三氯化钛金属配合物(1当量),室温反应16 h。反应结束后,用真空线将溶剂除去,残余物用二氯甲烷洗涤并通过硅藻土过滤,将滤液抽干,粗产品用二氯甲烷/正己烷重结晶,得到配合物。利用配体L1~L5与五甲基环戊二烯基三氯化钛金属配合物反应可以得到不同的茂金属配合物1~5。配合物的合成路线见图2。

图1 配体化合物合成路线Fig.1 Synthetic route of the ligands.
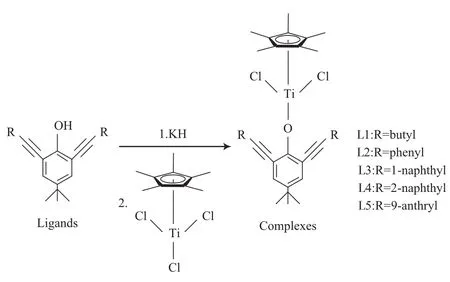
图2 茂金属配合物合成路线Fig.2 Synthetic route of the complexes.
2 结果与讨论
2.1 化合物性状、产率及1H NMR谱图
配体化合物L1(淡黄色油状物,产率68%):δ=7.26(s,2H),5.98(s,1H),2.48(t,J=7.0 Hz,4H),1.67~ 1.55(m,4H),1.55 ~ 1.44(m,4H),1.26(s,9H),0.95(t,J=7.2 Hz,6H);配合物1(红棕色粉末,产率98%):δ=7.24(s,2H),2.40(t,J=7.2 Hz,4H),2.24(s,15H),1.65~1.54(m,4H),1.52~1.39(m,4H),1.28(s,9H),0.94(t,J=7.3 Hz,6H)。
配体化合物L2(白色固体,产率72%):δ=7.62~7.55(m,4H),7.47(s,2H),7.41~7.34(m,6H), 6.13(s,1H),1.33(s,9H);配合物2(红棕色粉末,产率92%):δ=7.70~7.62(m,4H),7.47(s,2H),7.40~7.31(m,6H),2.19(s,15H),1.35(s,9H)。
配体化合物L3(黄色固体,产率57%):δ=8.56(t,J= 7.7 Hz,2H),7.94 ~ 7.82(m,6H),7.70~ 7.62(m,4H),7.58(t,J=8.0 Hz,2H),7.51(t,J=4.0 Hz,2H),6.40(s,1H),1.43(s,9H);配合物3(红棕色粉末,产率96%):δ=8.50(d,J= 8.5 Hz,2H),7.98(d,J=1.0 Hz,1H),7.96(d,J=1.0 Hz,1H),7.88(t,J=8.1 Hz,4H),7.67~7.60(m,4H),7.59~7.48(m,4H),2.14(s,15H),1.43(s,9H)。
配体化合物L4(淡黄色固体,产率52%):δ=8.11(s,2H),7.88~7.81(m,6H),7.64(d,J=1.5 Hz,1H),7.61(d,J=1.5 Hz,1H),7.55~7.49(m,6H),6.22(s,1H),1.36(s,9H);配合物4(红棕色粉末,产率95%):δ=8.23(s,2H),7.90~ 7.81(m,6H),7.74(d,J=1.5 Hz,1H),7.72(d,J=1.5 Hz,1H),7.56~7.48(m,6H),2.20(s,15H),1.39(s,9H)。
配体化合物L5(红棕色固体,产率75%):δ=8.75(d,J=8.6 Hz,4H),8.48(s,2H),8.05(d,J=8.4 Hz,4H),7.76(s,2H),7.65(t,J=7.1 Hz,4H),7.54(t,J=7.1 Hz,4H),6.57(s,1H),1.48(s,9H);配合物5(红棕色粉末,产率99%):δ=8.75(d,J=8.6 Hz,4H),8.49(s,2H),8.06(d,J=8.4 Hz,4H),7.80(s,2H),7.65(t,J=7.2 Hz,4H),7.56(t,J=7.5 Hz,4H),1.99(s,15H),1.48(s,9H)。
通过配体化合物与对应的配合物1H NMR谱图对比可以发现,增加了δ≈2.1的单峰,氢数为15,为环戊二烯基上五个甲基的氢;同时,相较于配体化合物,配合物1H NMR谱图中减少了δ≈6.2的单峰,该峰为配体中酚羟基的活泼氢,该峰的消除也证明了配体与金属的配位反应已发生。以L2为例给出了配体化合物合成对应配合物后的1H NMR谱图变化,见图3。
2.2 化合物的13C NMR谱图
配体化合物 L1:δ=154.8,142.7,129.1,109.7,96.1,75.4,34.0,31.3,30.9,22.1,19.3,13.6;配合物 1:δ=163.7,145.1,133.0,129.5,115.6,95.8,34.2,31.2,30.6,22.2,20.0,13.7,13.1。

图3 配体化合物L2与配合物2的1H NMR谱图Fig.3 1H NMR comparison of ligand 2(L2) and complex 2.Conditions:400 MHz,deuterated chloroform(CDCl3).
配体化合物L2:δ=154.9,143.3,131.7,130.0,128.7,128.5,122.8,109.5,94.9,84.0,34.2,31.3;配合物 2:δ=163.0,145.3,133.7,131.8,131.0,128.3,128.2,123.4,115.2,94.1,86.5,34.4,31.2,13.2。
配体化合物 L3:δ=155.3,143.5,133.3,130.7,130.0,129.3,128.4,127.1,126.6,126.3,125.3,120.5,109.9,93.3,89.0,34.3,31.4;配合物 3:δ=163.0,145.4,133.7,133.4,133.2,131.2,130.8,128.8,128.3,126.7,126.4,126.4,125.4,120.9,115.4,92.3,91.3,34.4,31.3,13.2。
配体化合物 L4:δ=155.0,143.4,133.0,133.0,131.7,130.1,128.3,128.2,127.9,127.8,126.9,126.7,120.1,109.5,95.4,84.4,34.3,31.4;配合物 4:δ=163.2,145.4,133.7,133.0,132.9,131.9,131.1,128.5,128.0,127.9,127.8,126.7,126.4,120.7,115.3,94.6,86.9,34.4,31.3,13.2。
配体化合物L5:δ=155.3,143.6,132.7,131.2,130.0,128.8,128.3,127.0,126.7,125.8,116.7,110.2,95.2,92.0,34.4,31.5;配合物 5:δ=162.9,145.3,133.5,132.7,131.9,131.2,128.6,127.6,126.5,125.8,117.4,115.5,97.1,91.2,34.5,31.4,13.1。
通过配体化合物与对应的配合物13C NMR谱图对比,在δ≈13.1,130处增加了两处碳谱峰,分别为五甲基环戊二烯基的甲基及环戊二烯基的碳,进一步证实了配体与金属的配位反应顺利进行,得到了配合物。以L1为例给出了配体化合物合成对应配合物后的13C NMR谱图变化,见图4。

图4 配体化合物L1与配合物1的13C NMR谱图Fig.4 13C NMR comparison of ligand 1(L1) and complex 1.Conditions:100 MHz,CDCl3.
2.3 化合物高分辨质谱
表1为化合物的HRMS谱图数据。由表1可知,HRMS谱图结果与计算结果匹配,进一步佐证了化合物的结构。同时,针对配体与五甲基环戊二烯基三氯化钛的配位反应,得到的配合物减少了一分子氯化氢的相对分子质量,证实了最后配位反应的发生。

表1 化合物的HRMS谱图数据Table 1 HRMS of the compounds
2.4 茂金属配合物的单晶X射线衍射结果
在二氯甲烷及正己烷的复合溶液中得到了配合物的单晶。其中配合物4得到了较好的晶体,单晶X射线衍射结果见图5。由图5可进一步确定配合物的结构。
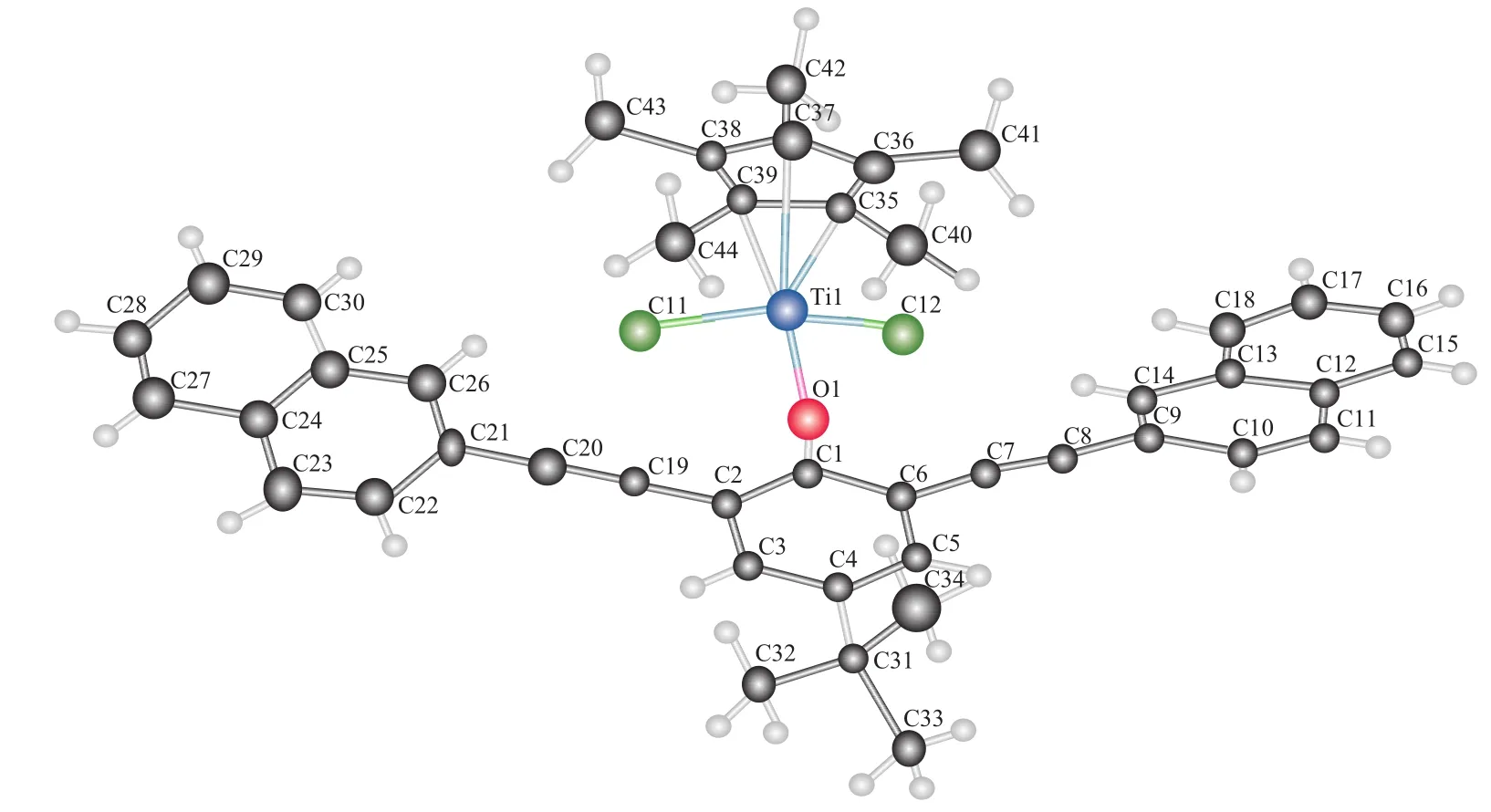
图5 配合物4的晶体结构Fig.5 Molecular structure of complex 4.
2.5 偶联反应的优化
在配体化合物的制备中,采用了对酚羟基的保护后偶联再脱保护的策略,增加了反应步骤。但如果直接进行偶联反应的话,反应效果较差,收率低于20%,原因可能是在钯的催化作用下,发生了进一步的关环反应,得到了香豆酮(图6)。当炔基的取代基R为叔丁基时,较大的位阻抑制了下一步关环反应的发生,可在不引入保护基的情况下直接偶联[19]。虽然通过减少反应时间可在一定程度上抑制关环反应的发生,但由于副产物的存在,增加了配体化合物分离纯化的难度。引入保护基后,虽然增加了反应步骤,但反应产率明显提高,且保护基的引入和脱除并不需要纯化,几乎是当量的反应,使配体的合成效率明显提升。
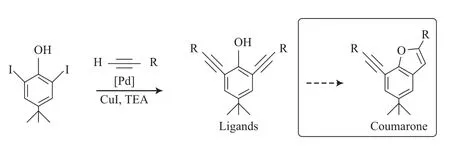
图6 一步法合成配体化合物Fig.6 One-step synthesis of the ligands.
3 结论
1)通过1H NMR,13C NMR,HRMS谱图及单晶X射线衍射的数据分析,证实了配体化合物及对应配合物的结构,并佐证了配位反应的顺利发生。
2)在配体合成过程中,保护基的引入在保证反应简洁度的同时,大大提高了反应效率,反应产率和分离效果得到明显提升。
3)通过简单的配位反应,合成了一种新型的邻位双炔基取代的苯酚类茂金属配合物。通过改变苯酚类配体炔基上的取代基,从而使配合物金属中心的电子云密度和空间位阻效应发生变化,进而得到不同性能的催化剂。在此研究的基础上,催化剂的应用及结构优化在进一步研究中。
