稀土对TiAl/Ti3Al界面稳定性和延性影响的第一性原理研究
2020-10-12苑光明董明慧王学文刘恩超
苑光明,董明慧,王学文,刘恩超
(齐鲁理工学院, 济南 250200)
随着人们对航空发动机的服役温度、轻量化提出更高的要求,传统的镍基合金已经不能满足设计的要求,因此寻找一种新的可以替代传统高温合金的新型材料已经迫在眉睫[1]。TiAl由于具有良好的高温机械性能和低密度等优点,在航空发动机、飞行器、船舶制造等领域得到广泛的应用[2]。根据Ti-Al二元合金相图可知Ti-Al凝固可以析出TiAl、Ti3Al、TiAl2、TiAl3四种主要的相。作为第二代Ti-Al合金,应用最广的是γ-TiAl和α2-Ti3Al。虽然γ-TiAl和α2-Ti3Al高温性能良好,但是低温塑性、韧性以及强度较差,制约了铸造成型和机械加工[4-5],因此如何提高室温机械性能成为人们关注的焦点问题。实验上关于两种结构的微观组织和结构性能已经得到了深入的研究,透射电镜研究发现在晶体学关系上以γ-TiAl[1-10](111)和α2-Ti3Al[11-20](0001)面最为常见[6]。相比单晶形式的γ-TiAl合金,γ-TiAl和α2-Ti3Al形成的双相层状结构具有更高的延展性和硬度,因此得到更广泛的关注[7]。
为了提高TiAl合金的综合机械性能,人们从理论和实验上做了大量探究工作。研究发现合金化是提高TiAl合金室温塑韧性以及强度的重要手段之一[8]。Li[9]通过第一性原理研究发现B掺杂TiAl/Ti3Al后能够提高界面的结合强度;Neelam[10]等研究发现Mo掺杂γ-TiAl能够很好的提高高温抗氧化性。Chen[11]等研究发现Nb分别替代TiAl2中的Ti原子和Al原子后形成能都是负值,结构稳定,且断裂韧性和塑性都得到很好的提高。王海燕[12]分别研究了V、Nb、Ta、Cr、Mo、W对γ-TiAl合金的力学性能的影响,发现Cr、Mo、W能明显提高γ-TiAl合金的韧性,且Mo含量在12.5%时韧性最强。对于γ-TiAl和α2-Ti3Al界面结构,刘东伟[13]分析了界面强韧化机理与电子结构间的关系,宋庆功[14]研究发现Mo、Cr能提高γ-TiAl和α2-Ti3Al层状结构界面的延性,但是V对于延性的影响较小。稀土元素对于金属材料力学性能的影响也非常明显,宋庆功等[15]采用密度泛函理论研究了La分别替换TiAl合金中的Ti和Al原子后的结构稳定性和延性,发现La 替换Ti和Al原子都能得到稳定的结构,但是随着掺杂量的增大稳定性下降。宋庆功[16]等研究发现Nb含量在1.85%~4.17%时γ-TiAl延性最好。虽然稀土对提高TiAl合金的延性有一定的影响,但是对TiAl/Ti3Al界面的影响还未见报道。因此本文将采用基于密度泛函理论的第一性原理研究常见的稀土La、Ce、Nd、Sm对TiAl/Ti3Al界面力学性能的影响,从电子层面探究稀土影响界面稳定性和力学性能的机理。
1 计算方法与模型
计算过程采用了基于密度泛函理论的第一性原理[17],以Ceperley-Alder-Perdew-Zunger (CAPZ)作为交换关联函数[18-19]。平面波截断能为500 eV,Monkhorst-Pack[20]k点网络密度选取的为4×4×4。自洽能量收敛标准小于1.0×10-5eV/atom。Ti电子组态:3s23p63d24s2,Al电子组态:3s23p1。
γ-TiAl空间群是P4/mmm,具有L10型面心立方结构;α2-Ti3Al空间群是属于P63/mmc,具有D019型密排六方结构。根据透射电镜发现的晶体学取向关系,以γ-TiAl[1-10](111)和α2-Ti3Al[11-20](0001)作为研究的模型晶界。由于γ-TiAl属于面心立方结构,故在(111)密排面上具有ABCABCABC堆垛,三层原子为一周期;α2-Ti3Al具有密排六方结构,(0001)密排面上具有ABABAB堆垛。为了使计算模型更加接近于实验样品,γ-TiAl选取了6层原子(2个周期),α2-Ti3Al选取了6层原子(3个周期)。
图1为建立的晶界模型,粉红色原子代表Al,灰色原子代表Ti。1、4代表远离界面处的原子,2、3代表界面处的原子。为了验证计算方法的可靠性,首先对γ-TiAl和α2-Ti3Al两个单胞进行几何结构优化,得到γ-TiAl晶格常数a=b=0.407 nm,c=0.406 nm;α2-Ti3Al晶格常数a=b=0.578 nm,c=0.464 nm。Wei[21]通过计算得到的γ-TiAl晶格常数a=b=0.399 nm,c=0.408 nm;α2-Ti3Al晶格常数a=b=0.574 nm,c=0.465 nm。Koizumi[22]通过实验测得的γ-TiAl晶格常数a=b=0.4 nm,c=0.408 nm;α2-Ti3Al晶格常数a=b=0.577 nm,c=0.462 nm。和其他理论与实验测定的结果相比可知计算得到的晶格常数差别小于2%,说明计算方案合理,因此四种稀土元素掺杂TiAl/Ti3Al界面也是采用的此计算方案。

图1 TiAl/ Ti3Al界面模型示意图
2 计算结果与讨论
2.1 能量稳定性
材料结构形成的难易程度可以用形成能表征,形成能为负说明是放热反应,结构稳定,反之则不稳定。形成能可由式(1)求得[23]。
Eform=(Et-mETi-nEAl-EX)/(m+n+1)
(1)
式中:Et是稀土掺杂后的总能量;ETi、EAl、EX分别是Ti、Al和稀土X(X=La、Ce、Nd、Sm)单质完全弛豫后的单原子能量;m和n分别是计算模型中的Ti和Al原子个数。
图2是稀土元素分别位于Al1、Al2、Al3、Al4和Ti1、Ti2、Ti3、Ti4位置处的形成能。计算结果显示所有掺杂后的形成能都是负值,是放热反应,结构稳定。稀土替换晶界处的Ti原子(Ti2、Ti3)和Al原子(Al2、Al3)的形成能要低于替换基体原子(Ti1、Ti4、Al1、Al4)的形成能,因此稀土元素容易在晶界处偏析。4种稀土掺杂后的形成能总体上满足关系ESm>ENd>ECe>ELa,这主要是由于稀土原子半径RSm>RNd>RCe>RLa造成的。掺杂原子半径增大必然导致晶格畸变,因此形成能随之增大。另外比较稀土替位掺杂同层的Ti和Al原子可知,La、Ce和Sm替位Al后的形成能要比替位Ti的低,因此La、Ce和Sm更倾向于替位Al原子,而Nd原子刚好相反,倾向于替位Ti原子。

图2 La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的形成能示意图
2.1 界面结合强度
晶界结合强弱可以由组成晶界的两个相分裂成单独的相时外界所做的功与界面面积的比值来表示,断裂功越大说明晶界结合越稳固,反之则容易断裂。断裂功可以由式(2)[24]计算出来。
Wsep=(Eγ-TiAl+Eα2-Ti3Al-Eγ-TiAl/α2-Ti3Al)/2A
(2)
其中:Eγ-TiAl和Eα2-Ti3Al是分立结构的总能量;Eγ-TiAl/α2-Ti3Al是界面结构的总能量;A是界面面积。
由于La、Ce、Nd和Sm在界面结构中更倾向于占据界面位置,因此仅计算了稀土原子替位Al2、Ti2、Al3和Ti3四处原子的断裂功。图3即为计算出来的断裂功,其中纯净的γ-TiAl和α2-Ti3Al界面断裂功为3.25 J/m2,La替位Al2、Ti2、Al3和Ti3后的断裂功分别为3.12 J/m2、3.18 J/m2、3.05 J/m2、3.09 J/m2,La在界面处的断裂功均小于纯净界面的断裂功,说明固溶于界面的La原子使得界面结合强度降低,有利于改善界面延性。Ce分别替位上述原子后的断裂功为3.06 J/m2、3.12 J/m2、3.28 J/m2、3.15 J/m2,其中Ce替位Al3处的原子得到的断裂功略高于纯净界面的断裂功,而替位其他三处原子均小于纯净界面。Nd掺杂后的断裂功分别为3.38 J/m2、3.42 J/m2、3.35 J/m2、3.48 J/m2,所有的断裂功都大于纯净界面的断裂功,结合强度增大,不利于改善界面延性。Sm掺杂后的断裂功为2.94 J/m2、3.1 J/m2、3.08J/m2、3.27 J/m2,其中Sm替位Ti3原子的断裂功略高于纯净界面的,而替位其他位置的断裂功均小于纯净界面的,有利于改善界面延性。对比位于同一层面的原子(Al2和Ti2,Al3和Ti3)可以看出,稀土原子替位Al原子位置的断裂功小于替位Ti原子的断裂功。

图3 La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的断裂功示意图
2.2 弹性模量
材料的硬度和变形能力与弹性模量有关,其中切变模量G是切应力与切应变之比,模量大则表明材料难以变形。体变模量B是材料受到四周压力后产生形变程度的度量,模量大则表明难以压缩。Pugh[25]发现切变模量G和体变模量B的比值G/B能够反应材料的延性,研究表明G/B=0.5是区分延性与脆性的重要依据,当G/B<0.5时延性好,当G/B>0.5脆性明显。图4是计算出的La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的弹性模量大小,(a)是体变模量B,(b)是切变模量G,(c)是切变模量G与体变模量B的比值。计算出的纯净的TiAl/Ti3Al界面体变模量为104 GPa,切变模量为54 GPa,G/B=0.52。稀土掺杂后体变模量除Ce和Sm替位Al2处的Al原子,La和Sm替换Al3处的Al原子外,其他替位掺杂都能够提高体变模量的大小。而稀土掺杂后对于切变模量的影响不是很明显,其中La、Ce和Sm替位Al2处的原子切变模量分别为47 GPa、48 GPa和37 GPa,Sm替位Ti3处的原子切变模量为49 GPa,La和Sm替位Al3处的原子切变模量分别为41 GPa、44 GPa,La替位Ti3处的原子切变模量为46 GPa,上述替位掺杂的切变模量都比纯净结构的小,有利于切变变形。图图3(c)的G/B是切变模量G与体变模量B的比值,能够在一定程度上反应出材料的延性。La、Ce、Nd和Sm替换Al2原子后的G/B分别为0.45、0.48、0.67和0.4,其中Nd替换Al2后的G/B比值最大,不利于界面延性,La、Ce替换Al2原子后的G/B基本在0.5左右,对延性影响不是很明显,Sm替换Al2原子后的G/B小于0.5,有利于改善界面延性。La、Ce、Nd和Sm替换Ti2原子后的G/B分别为0.48、0.47、0.71和0.45,Nd替换Ti2后的G/B比值最大,不利于延性,而La、Ce和Sm替换Ti2原子后的G/B基本在0.47左右,非常接近0.5,对延性的影响不是很明显。La、Ce、Nd和Sm替换Al3原子后的G/B分别为0.43、0.55、0.61和0.45,Nd替换Al3后的G/B比值虽然依然很大,但是相比替换Al2和Ti2两处原子已经明显减小,另外La替换Al3原子后的G/B基本在0.43,有利于延性,但是Ce和Sm则大于0.5,不利于界面延性。La、Ce、Nd和Sm替换Ti3原子后的G/B分别为0.41、0.44、0.65和0.55,Nd和Sm替换Ti3后的G/B比值大于0.5,不利于界面延性,La、Ce替换Ti3原子后的G/B小于0.5,有利于延性。总体分析,Nd替换所有原子的G/B比值都很大,且远大于0.5,非常不利于界面延性,而La、Ce和Sm除Ce替换Al3原子、Sm替换Ti3外G/B比值都小于0.5,尤其是La替换Ti3原子和Sm替换Al2原子的G/B比值相比纯净的TiAl/Ti3Al界面减小非常明显,有利于改善界面延性。

图4 La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的弹性模量示意图
2.3 电荷布居数
为了从电子层面上探究稀土掺杂对TiAl/Ti3Al界面延性的影响,表1列出了La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的Mulliken布居数。单独的Al原子最外层3p电子有1个电子未配对,Ti原子最外层3d电子有2个未配对电子,因此Al-Ti容易形成共价键。由于共价键具有一定的方向性,Greenberg[26]等认为,共价键的方向性是造成TiAl合金脆性的主要原因,因此减小共价键的成键电子数能很好的减小脆性、提高延性。
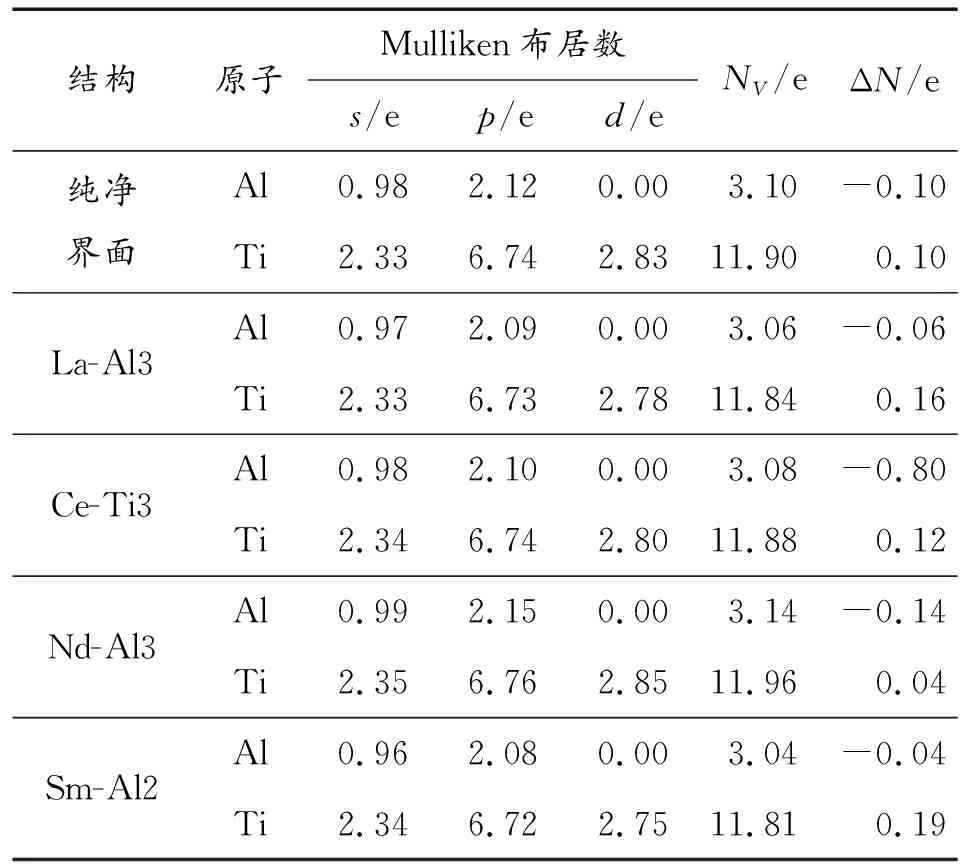
表1 La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的Mulliken布居数
表1中的s、p、d分别为不同的价电子态,NV是价电子总数,ΔN是原子得失电子数。由于掺杂体系中原子之间的距离不同,电子在原子之间分布也不尽相同,表1只是给出了原子核外价电子的平均分布数。从表1中可知,对于纯净的TiAl/Ti3Al界面而言,Al原子得到0.1个电子,而Ti原子失去0.1个电子。Al的3p电子数量为2.12个,Ti的3d电子数量为2.83个,因此Ti-Al之间p-d杂化电子总数为4.95个,杂化强度高,共价键电子多。La-Al3为La原子取代Al3处的原子结构,La取代Al3后Al的3p电子数量减小到2.09个,Ti的3d电子减小到2.78个,总的成键电子数量减小到4.87个,所有共价键强度减弱。同理,Ce取代Ti3处的原子总的成键电子数为4.9个,Sm取代Al2处的原子总的成键电子数为4.83个,均小于纯净结构的成键电子数,因此La、Ce、Sm能够减小共价键电子数。而Nd取代Al3原子后Al的3p电子却增加到2.15个,Ti的3d电子增加到2.85个,总的成键电子为5个,共价键成键强度增大,与图3断裂功和图4G/B得到的结论一致。
2.4 重叠布居数
表2所示为重叠布居数和键长的平均值。
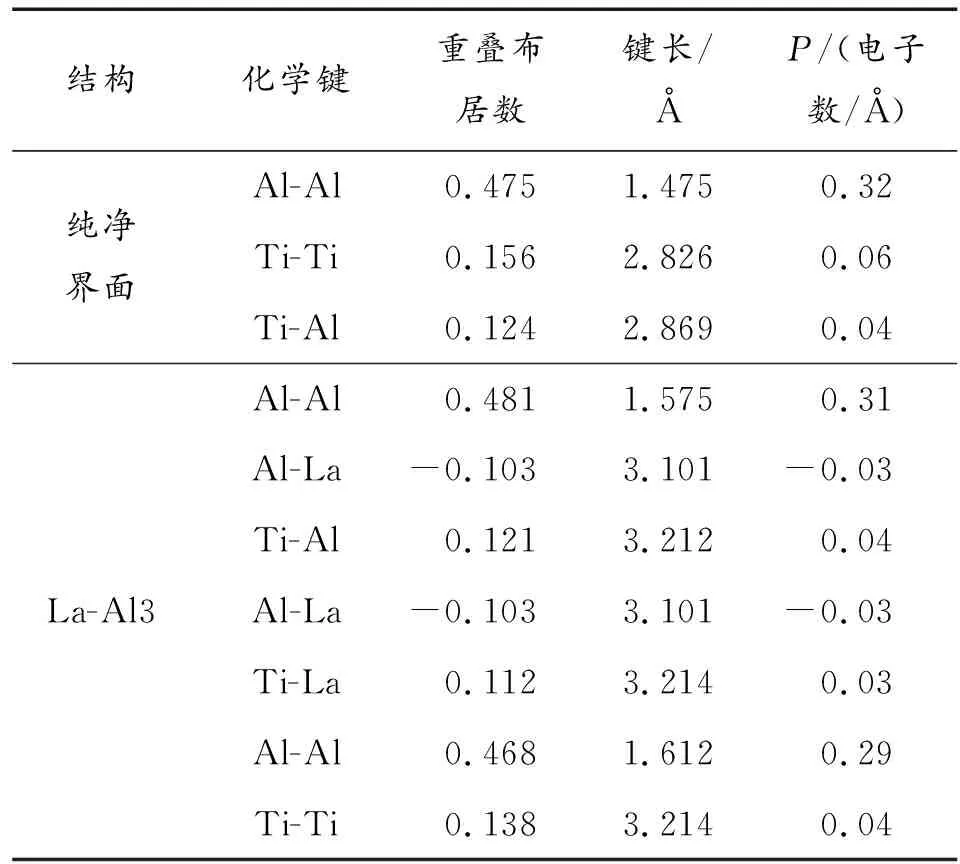
表2 La、Ce、Nd、Sm掺杂TiAl/Ti3Al界面的重叠布居数

续表(表2)
重叠布居数表示原子间电子云的重叠程度,常被用于描述原子之间的作用性质。重叠布居数为正时说明原子之间形成共价键,且数值越大共价键的强度越强,反之如果为负则说明原子相互排斥[15]。计算过程选取了4种稀土元素最为优先取向的结构模型。由于掺杂原子和Al、Ti原子的距离不同,所以表2中的重叠布居数和键长都是平均值。
在纯净的TiAl/Ti3Al以及稀土掺杂后的界面结构中,Al-Al键的P值最大,说明在共价键主要位于Al-Al之间。La替换Al3原子后Al-Al之间的P值由0.32减小到0.31,共价键强度降低,且Al-La之间P值为-0.03,说明Al-La相互排斥。Ce替换Ti3原子后Ti-Ce之间P值为-0.06,说明Ti-Ce相互排斥;Sm替换Al2后Ti-Sm之间P值为-0.07,说明Ti-Sm相互排斥。而Nd替换Al3后Al-Nd、Ti-Nd之间P值为正值,因此能够形成共价键,不利于改善界面的延性。
3 结论
1) La、Ce、Nd、Sm替位TiAl/Ti3Al晶界处原子的形成能比基体内的低,因此稀土元素更容易在晶界处偏聚,且四种稀土元素掺杂的形成能都为负值,因此形成稳定结构。
2) Ce替位Al3处原子、Sm替位Ti3处原子以及Nd替位所有界面原子的断裂功都高于纯净界面的断裂功3.25 J/m2,不利于改善界面延性;而替位其他位置处的原子断裂功都小于3.25 J/m2,有利于改善界面延性。
3) Ce替位Al3处原子、Sm替位Ti3处原子以及Nd替位所有界面原子的G/B都高于0.5,不利于改善界面延性;其他原子替位掺杂都小于0.5,有利于改善界面延性,与断裂功的结论一致。
4) Mulliken布居和重叠布局显示,Nd掺杂后的共价键强度增强,不利于界面延性改善,而La、Ce、Sm掺杂共价键强度减小,有利于改善界面延性。
