胶束电动色谱法测定五维他口服溶液中维生素B1、B2、 B6、烟酰胺、泛酸钙与苯甲酸钠
2020-09-29李丹凤黄罗健朱健萍卢日刚
李丹凤,黄罗健,朱健萍,卢日刚*
(1.广西壮族自治区食品药品检验所,广西 南宁 530021;2.桂林医学院 药学院,广西 桂林 541100)
五维他口服溶液是由维生素B1、B2、B6、烟酰胺、泛酸钙等成分组成的复方维生素B族口服制剂,主要用于厌食、营养不良、脚气病、糙皮病及缺乏维生素B族所致的各种疾患的辅助治疗[1],不良反应主要有呕吐、腹泻、便秘、皮疹、瘙痒、嗜睡、舌麻痹、乏力和呼吸困难等[2]。该制剂属于多剂量包装的口服溶液,处方中含有蔗糖,在保存或多次服用过程中易霉变,因此需加入防腐剂苯甲酸钠。但苯甲酸钠属于酸性物质,食用太多会增加机体的酸度,从而导致人体内碘、铁、钙等物质过多消耗与流失,且会对人的神经系统造成损害[3-4]。现行国家药品标准WS-1001-(HD-0408)-2002[5]采用高效液相色谱法(HPLC)测定五维他口服溶液中B1、B2、B6、烟酰胺的含量,但未对其处方中标注的泛酸钙和苯甲酸钠进行测定。泛酸钙又名维生素B5,是五维他口服溶液中起药理作用的主要成分之一。苯甲酸钠属于防腐剂,其含量涉及用药安全,必须建立相关含量的测定方法。
毛细管电泳技术是以高压电场为驱动力,以毛细管为分离通道,依据样品中各组分之间浓度和分配行为上的差异,从而实现分离的一类液相分离技术[6],已被广泛应用于苯甲酸、山梨酸、土霉素、人工合成色素和防腐剂等多种物质的测定[7-10]。胶束电动色谱(Micellar electrokinetic chromatography,MEKC)是目前应用较为广泛的一种毛细管电泳分离技术,可用于分离中性溶质以及带电溶质。该方法通过在缓冲液中加入离子型表面活性剂如十二烷基硫酸钠(SDS)而形成胶束,由于分析物在溶液和胶束间的分配作用及自身的泳动淌度不同,从而实现聚焦和分离[11],因此可适用于消栓通络片、雷公藤、复方氨酚烷胺胶囊等药物中有效成分的测定[12-16]。本研究根据维生素B1、B2、B6、烟酰胺、泛酸钙5个主药与防腐剂苯甲酸钠的性质,建立了胶束电动色谱法同时测定五维他口服溶液中此6个成分的含量,方法灵敏度高、重复性好、操作简便可靠,可用于该制剂的质量控制。
1 实验部分
1.1 仪器、试剂与材料
7100毛细管电泳仪,配二极管阵列检测器(美国Agilent公司);XD205DU电子天平(感量0.1 mg,梅特勒公司);Milli-Q 3800超纯水机(美国密理博公司);未涂渍的标准熔融石英毛细管柱(75 μm×50 cm,有效长度42 cm);维生素B1、B2、B6、烟酰胺、泛酸钙、苯甲酸钠对照品(纯度>99.5%,中国食品药品检定研究院);硼砂、硼酸、十二烷基硫酸钠(分析纯,国药集团化学试剂有限公司);乙腈(色谱纯,默克公司);实验用水为超纯水(电导率小于0.1 μS/cm);五维他口服溶液(广西婵方药业股份有限公司,简称GXCF,批号:191001、190302;广东南国药业有限公司,简称GDNG,批号:190405、190709;江苏神华药业有限公司,简称JSSH,批号:190322、190323)。
1.2 溶液配制
1.2.1 对照品溶液的配制精密称取维生素B1、B2、B6、烟酰胺与泛酸钙对照品各60、12、18、60、15 mg,分别置于20、20、20、20、50 mL棕色容量瓶中,加20%乙腈水溶解并定容至刻度,摇匀,即得单标储备液。精密称取苯甲酸钠对照品30 mg,置于10 mL棕色量瓶中,加入少量20%乙腈水溶解,再精密加入维生素B1、B2、B6、烟酰胺、泛酸钙的对照品储备液各1 mL,再用20%乙腈水定容至刻度,摇匀,即得维生素B1、B2、B6、烟酰胺、泛酸钙及苯钾酸钠的浓度分别为300、60、90、300、30、3 000 mg/L的混合对照品储备液。
混合标准工作液:取混合对照品储备液1.0、1.0、1.0、2.0、4.0 mL,分别置于50、20、10、10、10 mL棕色容量瓶中,加20%乙腈水稀释至刻度,得维生素B1与烟酰胺质量浓度为6.0、15、30、60、120 mg/L,维生素B2为1.2、3.0、6.0、12、24 mg/L,维生素B6为1.8、4.5、9.0、18、36 mg/L,泛酸钙为0.6、1.5、3.0、6.0、12 mg/L,苯甲酸钠为60、150、300、600、1 200 mg/L的系列混合标准工作液。
精密量取混合对照品储备液2 mL置于20 mL棕色量瓶中,加20%乙腈水稀释至刻度,摇匀,即得对照品溶液,过0.45 μm滤膜,超声脱气后于4 ℃避光保存,备用,用于精密度计算。
1.2.2 样品溶液的配制精密量取五维他口服溶液2 mL置于20 mL棕色量瓶中,加20%乙腈水定容至刻度,摇匀,过0.45 μm滤膜,超声脱气后于4 ℃避光保存,备用。
1.2.3 阴性样品溶液的配制按五维他口服溶液的处方配比,称取蔗糖2.0 g、橙皮酊1.0 g、乙醇2 mL、枸橼酸1.5 g置于100 mL容量瓶中,加水溶解并稀释至刻度,摇匀,制成阴性样品溶液。精密量取阴性样品溶液2 mL置于20 mL棕色容量瓶中,加20%乙腈水定容至刻度,摇匀,过0.45 μm滤膜,超声脱气后于4 ℃避光保存,备用。
1.3 电泳分离条件
以未涂渍的标准熔融石英毛细管柱(75 μm×50 cm,有效长度42 cm)为分离通道;40 mmol/L硼酸+40 mmol/L硼砂+20 mmol/L 十二烷基硫酸钠(pH 9.0)为分离缓冲液;分离电压为20 kV;电动进样:进样电压10 kV,进样时间为10 s;柱温25 ℃;维生素B1、B2、B6、烟酰胺和苯甲酸钠的检测波长为270 nm;在8.8~10.0 min时切换波长为200 nm,用于检测泛酸钙。
新毛细管使用前用1 mol/L氢氧化钠冲洗30 min,再用超纯水冲洗30 min,使其活化。每次进样前分别用0.1 mol/L氢氧化钠、超纯水、40 mmol/L硼砂+40 mmol/L硼酸+20 mmol/L SDS缓冲液(pH 9.0)各冲洗毛细管柱5 min,当天分析结束后用1 mol/L氢氧化钠冲洗30 min,再用超纯水冲洗30 min。同一缓冲液在进样5次后必须进行更换以保证分析物的保留时间稳定。上述溶液及试剂使用前均经0.45 μm滤膜滤过,并超声脱气。
2 结果与讨论
2.1 电泳分离条件的优化
2.1.1 分离缓冲液的优化缓冲溶液pH值会影响毛细管内壁的Zeta电位和电渗流,同时改变被测组分在毛细管内的电离状态,从而影响被测组分的迁移时间[12]。实验以40 mmol/L硼砂(pH值为9.3)添加不同体积40 mmol/L硼酸溶液配制不同pH值(7.5、8.0、8.5、9.0、9.3)的缓冲液,考察缓冲液pH值对待测组分峰形及分离度的影响。结果显示,pH值对各组分间的分离度和峰形影响较大,pH值为7.5时不能有效分离维生素B6和烟酰胺,且苯甲酸钠峰形较差;pH值升至8.0时,维生素B6和烟酰胺的分离度良好,但苯甲酸钠峰形仍得不到改善;pH值为8.5~9.3时,各化合物的峰形及分离度均较好。另外,实验发现同浓度硼砂与硼酸等比例混合时pH值恰好均为9.0,综合考虑各化合物的分离度、峰形、迁移时间以及操作便捷性,选择缓冲液pH值为9.0。
缓冲液浓度也会影响待测组分的峰形及保留时间,因此实验考察了终浓度均为20、30、40、50、60 mmol/L的硼砂+硼酸溶液对待测物分离度及峰形的影响。结果发现,缓冲液浓度对待测物峰形影响不大,各峰间的分离度较好,待测物的保留时间随缓冲溶液浓度的增加而增大。当缓冲液浓度大于40 mmol/L时,焦耳热增大,导致基线噪音增大,且分析物迁移时间变长,不利于分析。因此选择40 mmol/L硼砂+40 mmol/L硼酸作为缓冲液。在此条件下,维生素B6的峰形仍较差,出现了平顶峰,不利于分析,当向缓冲液中加入一定量十二烷基硫酸钠(SDS)可显著改善维生素B6峰形,因此考察了不同浓度SDS对待测物分离度及峰形的影响。结果显示,6种化合物的峰形不受SDS浓度的影响,但出峰时间随SDS浓度的增大而延长,分离度也相应增大。当SDS终浓度大于20 mmol/L后,苯甲酸钠与维生素B1的出峰时间均大于20 min。
综上,考虑分离度及分析时间等因素,最终选择40 mmol/L硼砂+40 mmol/L硼酸溶液+20 mmol/L SDS(pH 9.0)为分离缓冲液。
2.1.2 分离电压对分离效果的影响分离电压越高,分析物的迁移时间越短,因此考察了不同分离电压(10、15、20、25 kV)的分离效果。结果显示,10~25 kV分离电压下6种化合物的分离效果均良好,但低电压(10、15 kV)下的分离时间较长,超过20 min;20 kV和25 kV时的分离时间较短,可在10 min内出峰,但高电压下毛细管中产生的焦耳热会随之增加,从而会引起峰形展宽,因此选择最佳分离电压为20 kV。
2.1.3 检测波长的选择通过6个化合物在200~400 nm范围内全波长扫描图发现,维生素B1、B2、B6、烟酰胺在270 nm附近有最大吸收,苯甲酸钠虽然在225 nm附近有最大吸收,但270 nm处的吸收值也很大,为便于检测选择270 nm作为维生素B1、B2、B6、烟酰胺和苯甲酸钠的检测波长。泛酸钙在大于220 nm波长时无紫外吸收,仅在低波长处有末端吸收,因此选择200 nm为其测定波长。泛酸钙的出峰时间在9 min左右,因此选择在8.8~10.0 min时将波长切换为200 nm。

图1 混合标准溶液的电泳图Fig.1 Electrophoretogram of the mixed solution1.vitamin B2,2.vitamin B6,3.nicotinamide,4.calcium pantothenate,5.sodium benzoate,6.vitamin B1
综上,优化后维生素B1、B2、B6、烟酰胺、苯甲酸钠、泛酸钙的电泳条件见“1.3”所述,其电泳图见图1,由图可见,6种组分可在13 min内完全分离,分离度较好。
2.2 方法学验证
2.2.1 专属性试验五维他口服溶液中的其他辅料成分如橙皮酊、乙醇和枸橼酸可能会对其6种组分的测定产生干扰,因此实验按其处方配比称取相应的辅料成分,按照“1.2.3”方法制成阴性空白样品溶液,在优化条件下进样分析。结果显示,阴性样品溶液中其他组分不影响待测组分的测定,方法的专属性良好。
2.2.2 标准曲线、线性范围与检出限分别精密量取“1.2.1”配制的系列混合标准工作液,在优化条件下进样测定,重复进样3次,以6个化合物的3次峰面积的平均值(y,AU)对其质量浓度(x,mg/L)进行线性回归,以3倍信噪比(S/N=3)计算方法的检出限(LOD),以S/N=10计算方法的定量下限(LOQ)。结果显示,维生素B1、B2、B6、烟酰胺、苯甲酸钠、泛酸钙在一定质量浓度范围内线性良好,相关系数(r)不低于0.998 9,LOD为0.012~0.250 mg/L,LOQ为0.042~0.830 mg/L(表1)。
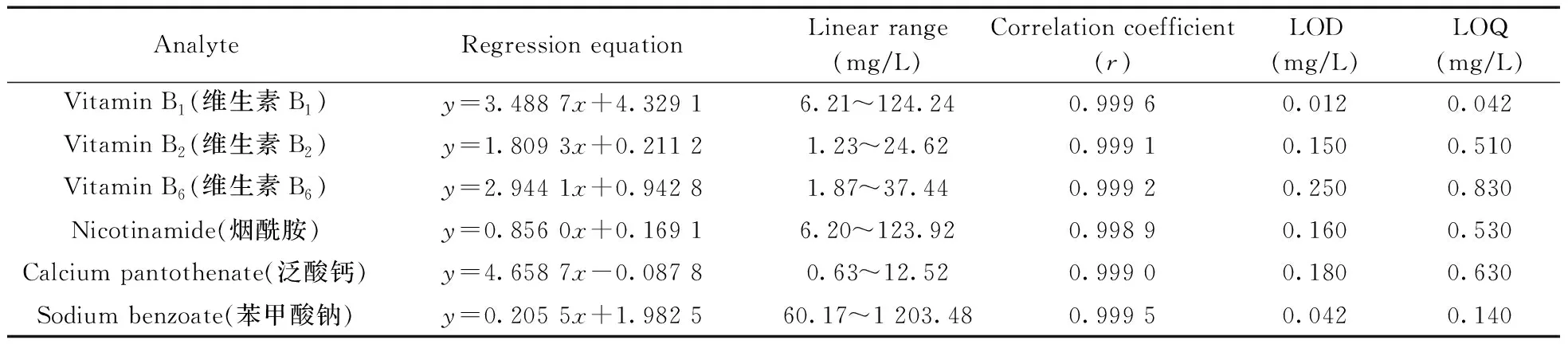
表1 6 个化合物的线性方程、线性范围、相关系数(r)、检出限和定量下限Table 1 Regression equations,linear ranges,correlation coefficients(r),LODs and LOQs of six compounds
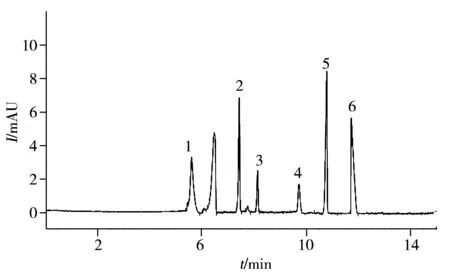
图2 样品溶液的电泳图Fig.2 Electrophoretogram of the sample solution1.vitamin B2,2.vitamin B6,3.nicotinamide,4.calcium pantothenate,5.sodium benzoate,6.vitamin B1
2.2.3 重复性与稳定性取五维他口服溶液(批号:191001)按“1.2.2”方法平行制备6份样品溶液,在优化条件下测定,重复进样3次并记录6个待测组分的平均峰面积和迁移时间。结果显示,维生素B1、B2、B6、烟酰胺、泛酸钙及苯甲酸钠的峰面积及迁移时间的相对标准偏差(RSD,n=6)均小于2.0%,表明方法的重复性良好。
取五维他口服溶液(批号:191001)按“1.2.2”方法配制样品溶液,分别在0、2、4、8、16、24、48、72、96、120 h时进样测定。结果显示,在24 h内6个成分峰面积的RSD均小于2.0%,而放置24 ~120 h后,6个成分峰面积的RSD为4.5%~18%,表明待测组分在24 h内稳定,超过24 h后不稳定,因此建议现用现配。
2.2.4 加标回收率与相对标准偏差取批号191001的五维他口服溶液,向其中添加一定量“1.2.1”配制的混合对照品溶液,配成低、中、高3个浓度的加标溶液,在优化条件下重复进样3次,记录平均峰面积,计算待测组分的回收率与RSD。结果显示,维生素B1、B2、B6、烟酰胺、泛酸钙及苯甲酸钠的回收率为97.0%~103%,RSD为0.60%~1.8%,表明方法的准确度高、精密度好,能够满足五维他口服溶液中此6个成分的同时测定要求。
2.3 实际样品测定
采用本文建立的MEKC方法对来自3个厂家6个不同批次的五维他口服溶液进行检测,每个样品平行3份,在优化条件下测定维生素B1、B2、B6、烟酰胺、泛酸钙及苯甲酸钠的含量(表2)。结果显示,6种化合物的测定结果与标示值相差不大,均在质量标准规定限度的90.0%~110%范围内,表明方法准确性较好,可用于五维他口服溶液的质量控制。图2为批号191001样品的电泳图。
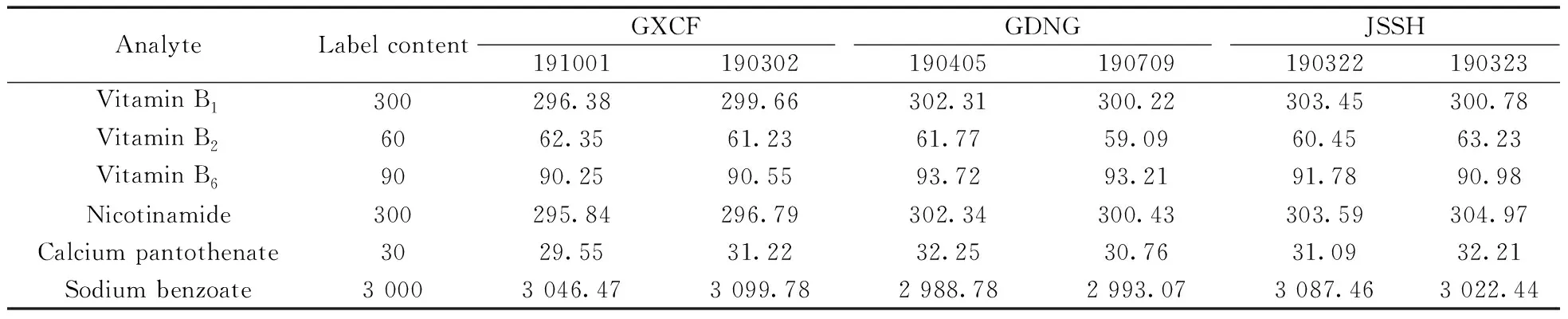
表2 实际样品的测定结果(n=3)Table 2 Analytical results for the real samples(n=3) ρ/(mg·L-1)
3 结 论
本文建立了胶束电动色谱法同时测定五维他口服溶液中维生素B1、B2、B6、烟酰胺、泛酸钙和苯甲酸钠含量的分析方法,该方法操作简便可靠,灵敏度高、重复性好,具有很好的实际应用价值,可用于该制剂的质量控制。
