通过式高效净化/超高效液相色谱-串联质谱法测定化妆品中73种糖皮质激素
2020-09-29杨飘飘李丽霞
刘 红,杨飘飘,李丽霞
(1.湖北省药品监督检验研究院,湖北 武汉 430075;2.湖北省药品质量检测与控制工程技术研究中心,湖北 武汉 430075)
糖皮质激素类药物由Kendall于1935年首次发现,20世纪50年代以来,许多肾上腺糖皮质激素类药物被合成,其核心结构为环戊烷多氢菲核[1]。该类药物可抑制纤维细胞增生,减少5-羟色胺形成,因而对皮肤有一定的嫩白作用[2]。为谋取高额利润,一些不法商家在化妆品中违法添加糖皮质激素,然而长期滥用糖皮质激素可引起激素依懒性皮炎[3],临床表现为皮肤明显变薄、毛细管扩张、痤疮和色素沉着等症状。《化妆品安全技术规范》(2015年版)规定糖皮质激素为禁用组分[4],涉及到49种糖皮质激素;GB/T 24800.2-2009涵盖41种糖皮质激素[5]。但为规避监管,仍有不法商家非法添加标准方法之外的糖皮质激素[6]。因此,研究出一种同时测定化妆品中更多种糖皮质激素、更易操作、准确定性定量的检测方法尤为重要。
目前糖皮质激素类药物的检测手段主要有薄层色谱法[5]、高效液相色谱法[7-9]、高效液相色谱-串联三重四极杆质谱法[10-13]和高效液相色谱-高分辨质谱法[14-15]等,但薄层色谱法常用于定性筛查,无法准确定量;高效液相色谱-串联三重四极杆质谱法在定量方面的灵敏度优于高效液相色谱法和高效液相色谱-高分辨质谱法,故应用最为普遍。由于化妆品基质复杂,目标物结构相似,目前报道的前处理方法有固相萃取法[5,16-17]或QuEChERS法[18],但这些方法具有步骤繁琐、耗时等不足。
本研究针对化妆品基质复杂、以油性原料和水为主的特性,采用通过式高效净化除去脂溶性物质,净化效率高,操作简便,结合超高效液相色谱-串联质谱(UPLC-MS/MS)法对水剂、乳液、膏霜3类基质化妆品中73种糖皮质激素进行了测定。方法快速、简单、稳定,结果可靠,能够满足化妆品中糖皮质激素风险筛查及监测工作的需要。
1 实验部分
1.1 仪器、试剂与材料
Waters Acquity UPLC/XEVOTMTQ-S 液相色谱-三重四极杆串联质谱仪(美国 Waters 公司);XP504电子天平(瑞士梅特勒公司);Milli-Q 超纯水器(美国Millipore 公司);LC-250超声波清洗机(山东济宁鲁超超声设备有限公司);台式离心机(德国Thermo Biofuge公司)。Waters CORTECS C18(150 mm×2.1 mm,2.7 μm,美国 Waters 公司);固相萃取柱为Oasis®PRiME HLB(200 mg/6 mL,60 mg/3 mL,美国Waters公司);甲醇、乙腈(色谱纯,默克股份两合公司);甲酸铵、甲酸(色谱纯,Aladdin 公司);乙酸铵(色谱纯,Macklin公司)。
73种糖皮质激素标准物质(见表1)分别购自中国食品药品检定研究院、欧洲药品质量管理局、美国药典委员会、加拿大 TRC 公司、美国Stanford 分析化学公司、德国 Dr.Ehrenstorfer 公司、美国A ChemTek公司。
1.2 对照溶液的配制
精密称取各对照品适量置于10 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得100 μg/mL的各单标对照储备溶液。分别精密量取适量各单标对照储备溶液于同一10 mL容量瓶中,加甲醇稀释至刻度,摇匀,即得200 ng/mL的混合标准储备液。
1.3 样品制备
称取均匀试样约0.2 g,置于15 mL离心管中,加入饱和氯化钠溶液3 mL,涡旋30 s,分散均匀,精密加入4 mL乙腈,涡旋30 s,超声提取30 min,涡旋混合摇匀,取上清液过PRiME HLB柱,重力作用下收集全部流出液。经0.22 μm滤膜过滤,滤液供UPLC-MS/MS测定。
1.4 色谱条件
色谱柱:Waters Cortecs C18(150 mm×2.1 mm,2.7 μm);柱温:35 ℃;流速:0.3 mL·min-1;进样体积:2 μL;流动相:乙腈(A)和0.1%乙酸溶液(B)。梯度洗脱程序:0~6 min,5%~15% A;6~16.8 min,15%~23.4% A;16.8~18.8 min,23.4% A;18.8~38 min,23.4%~42% A;38~44 min,42%~67.8% A;44~46 min,67.8% A;46~50 min,67.8%~85% A;50~52 min,85%~5% A;52~56 min,5% A。
1.5 质谱条件
离子源:电喷雾离子源;扫描方式:正离子扫描;检测方式:多反应监测(MRM);去溶剂气流速:800 L/h;去溶剂温度:350 ℃;毛细管电压:3.2 kV;73种待测物的质谱参数见表1。
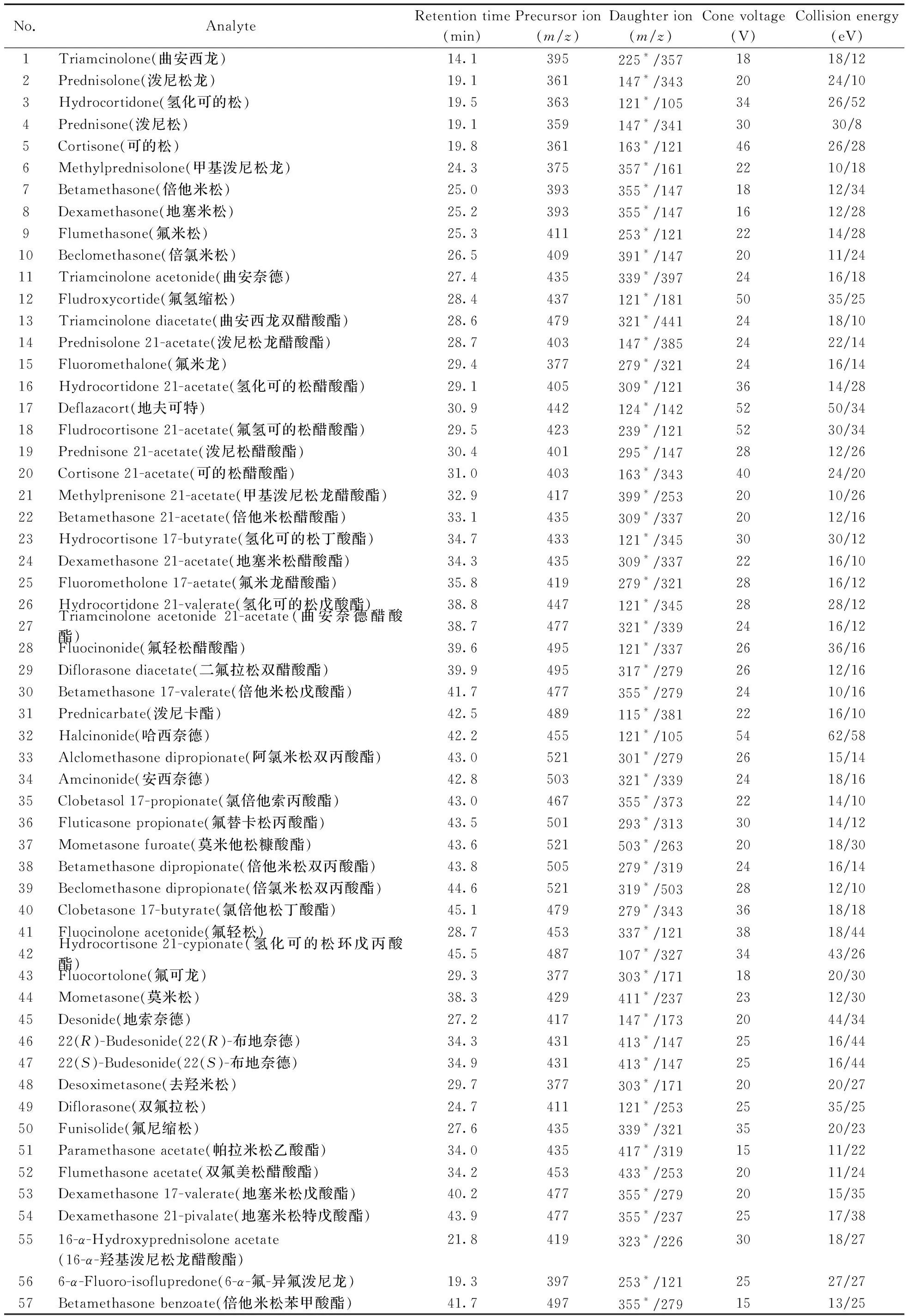
表1 73种分析物的质谱参数Table 1 MS parameters of the 73 analytes
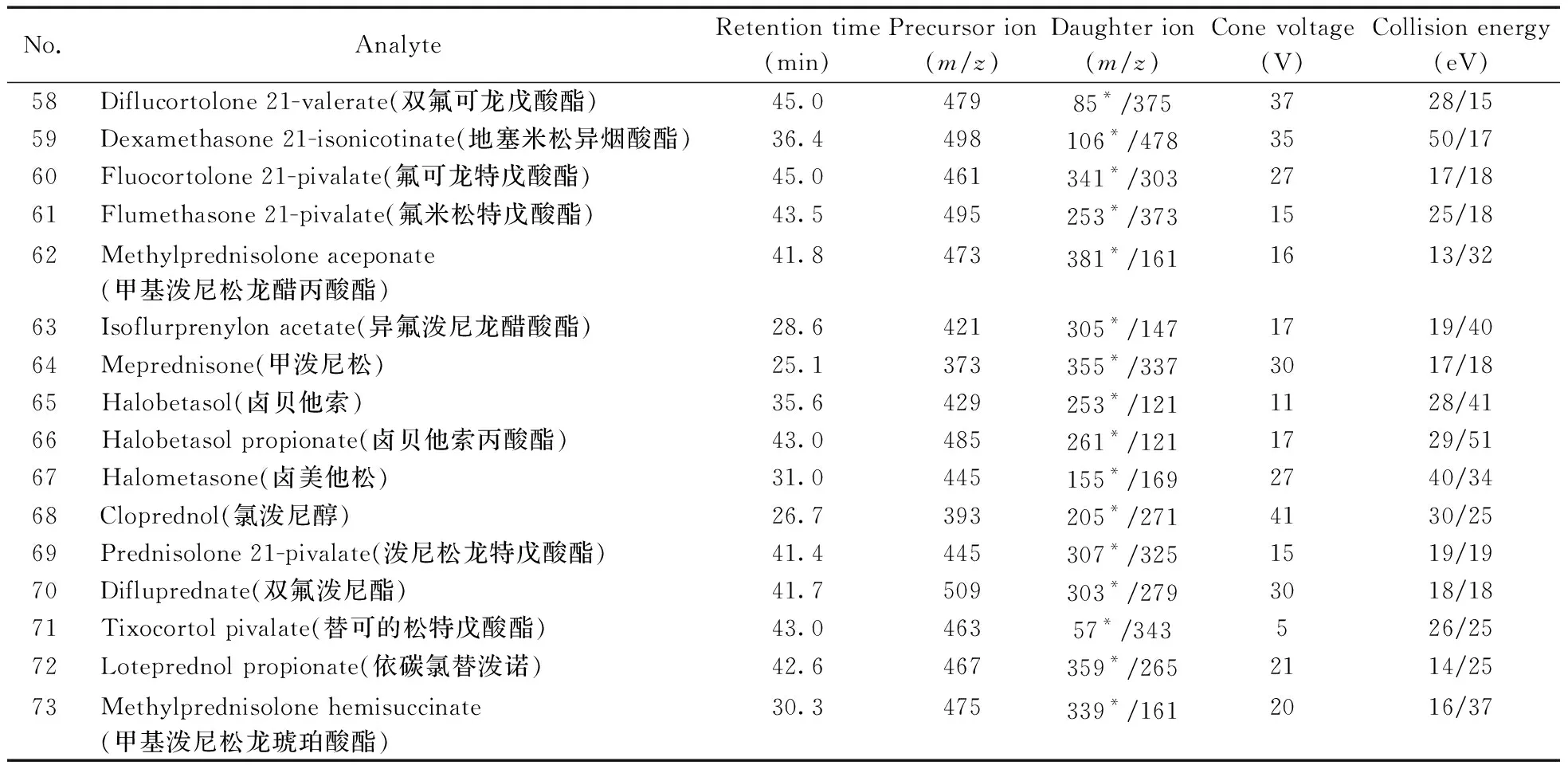
(续表1)
2 结果与讨论
2.1 样品提取方法的优化
糖皮质激素为甾体化合物,其结构特征是在固醇核D环的C17上有α羟基,在C环的C11上有氧或羟基[19]。因此,糖皮质激素类化合物多属于弱极性或中等极性,文献[16-17]以乙腈或甲醇为提取溶剂。考虑到乙腈作为提取溶剂更易分层,不易乳化,故选用乙腈为提取溶剂。化妆品基质复杂,由油性、粉质、溶剂类、胶质、表面活性剂、香精香料、颜料色素、保湿剂、防晒剂、防腐剂和抗氧化剂等原料根据不同的产品需求组合而成[20],所以分散剂的选择至关重要。实验比较了以水、饱和氯化钠为分散剂时73种糖皮质激素的回收率。结果表明,饱和氯化钠作为分散剂时,有73%(53/73)的目标物回收率更接近100%,而水作为分散剂时,仅有27%(20/73)的目标物回收率更接近100%,分析原因可能为饱和氯化钠属强电解质,作为分散剂和破乳剂可增加样品的表面张力,使分散更完全。所以最终选择饱和氯化钠作为分散剂。
2.2 色谱条件的优化
糖皮质激素以环戊烷多氢菲核为母核,仅取代基位置不同,本研究中共有12组同分异构体,如22(R)-布地奈德和22(S)-布地奈德等。采用0.1%乙酸溶液和乙腈为流动相,比较了Agilent Poroshell EC-C18(100 mm×2.1 mm,2.7 μm) 、Waters Cortecs C18(150 mm×2.1 mm,2.7 μm)和Waters BEH C18(100 mm×2.1 mm,1.7 μm)3种色谱柱对目标化合物分离效果和峰形的影响。结果表明,73种药物在3种色谱柱上的峰形均较好,但分离效果差异较大,12组同分异构体在Agilent Poroshell EC-C18(100 mm×2.1 mm,2.7 μm) 和Waters BEH C18(100 mm×2.1 mm,1.7 μm)上不能同时分离,而在Waters Cortecs C18(150 mm×2.1 mm,2.7 μm)上能实现较好分离,因此本文选用Cortecs C18(150 mm×2.1 mm,2.7 μm)色谱柱。
考察了乙腈-0.1%乙酸溶液、乙腈-0.1%甲酸溶液、乙腈-0.01 mol·L-1甲酸铵和乙腈-0.01 mol·L-1乙酸铵4种流动相体系对目标化合物分离效果和信号强度的影响。结果显示,上述4种流动相体系下,73种糖皮质激素的分离度和保留时间相似,但以乙腈-0.1%乙酸溶液作为流动相时的峰形最好,且绝大部分目标化合物的响应优于其他流动相体系。故选择乙腈-0.1%乙酸溶液作为流动相。

图1 典型同分异构体的基峰色谱图Fig.1 Base peak chromatogram of typical isomersthe peak numbers denoted were the same as those in Table 1
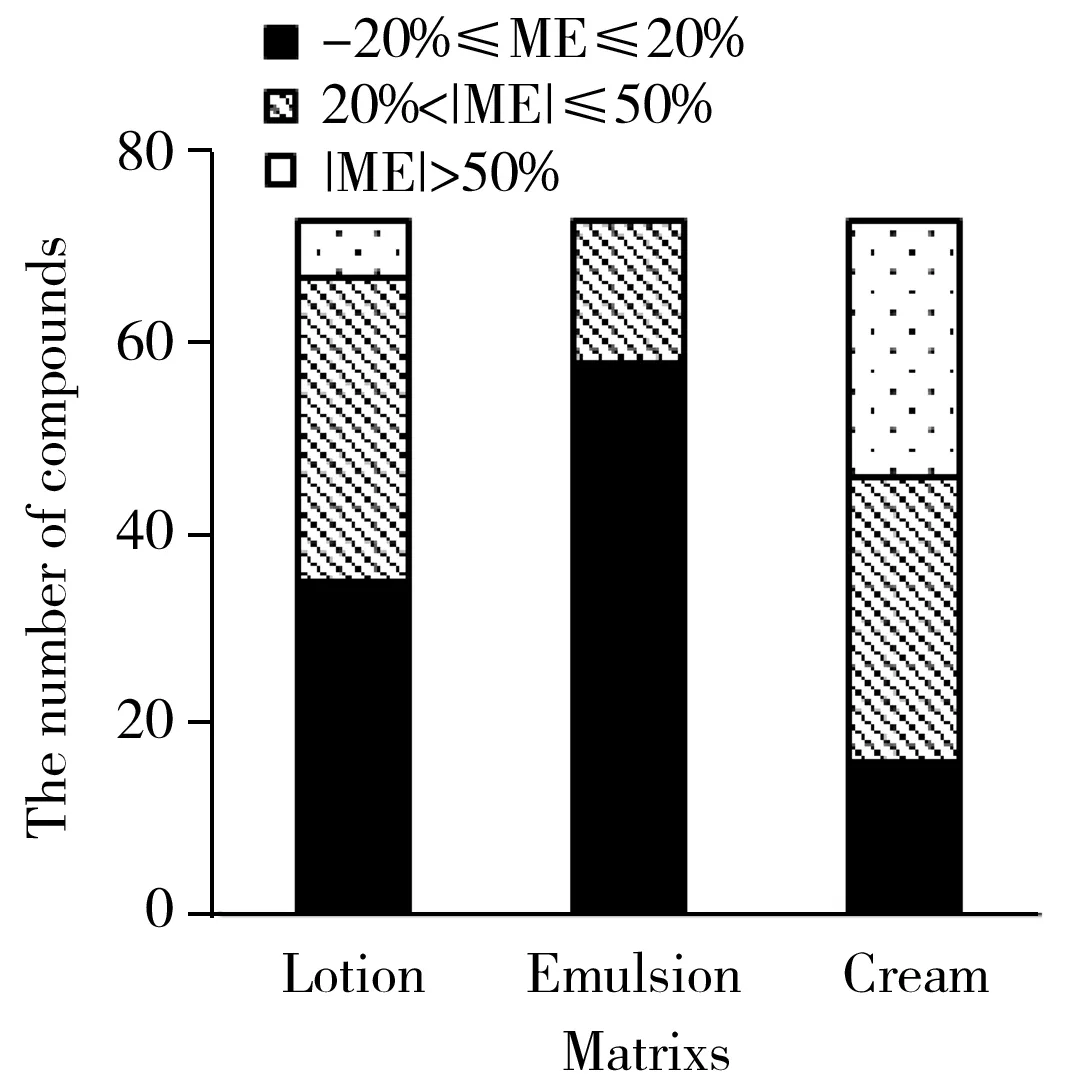
图2 3种基质化妆品基质效应的比较Fig.2 The comparison of matrix effect for three matrices
2.3 质谱条件的优化
在电喷雾离子源下,分别对质量浓度为200 ng/mL的目标化合物对照溶液做正离子和负离子全扫描,发现73种目标化合物均在正离子模式下获得响应最佳的准分子离子峰。由于糖皮质激素有相同的母核,73种目标化合物中有12组同分异构体,而且有些化合物虽不是同分异构体,但其质量数相同,裂解规律相似,易产生相同的碎片,所以选择特征的、无干扰且稳定的子离子对作为定量定性离子,以多反应监测模式优化各质谱参数,使化合物的准分子离子与特征碎片离子产生的离子对强度达到最大。优化条件下典型同分异构体的色谱图见图1。
2.4 净化条件的优化
样品经乙腈萃取后的溶液中含有大量脂溶性物质[21],因此利用固相萃取柱净化样品溶液。有文献采用HLB固相萃取柱净化[5,16-17],但步骤繁琐、耗时较长。而新型PRiME HLB 柱属于过滤型固相萃取柱,无需活化和平衡,能有效吸附脂溶性干扰物,过程简单、高效。因此,本实验选取200 mg/6 mL和60 mg/3 mL两种型号的PRiME HLB固相萃取柱进行了比较。结果显示,与过柱前相比,净化后的提取液澄清度均有显著提升。对两柱的提取回收率进行统计学t检验,结果表明两者有显著性差异,60 mg/3 mL固相萃取柱的平均提取回收率更好,故选择PRiME HLB(60 mg/3 mL)固相萃取柱进行净化。
2.5 基质效应
基质效应(ME)是由于样品在离子化时基质成分与目标化合物相互竞争电离所致,包括基质增强效应和基质抑制效应。采用ME=(1-基质对照溶液的峰面积/溶剂对照溶液的峰面积)×100%对73 种化合物的基质效应进行评价。当-20%≤ME≤20%时为弱基质效应;当 20%<|ME|≤50% 时为中等基质效应;当 |ME|>50% 时为强基质效应[22];通常采用基质标准曲线计算以获得更真实的样品数据[23]。本研究对水剂、乳液、膏霜3类化妆品的基质效应进行考察,由图2可知,膏霜类化妆品受基质影响较大,水剂次之,乳液类化妆品受基质效应影响最小。为了能更准确地测定目标化合物,本实验采用基质匹配标准曲线消除基质效应。
2.6 方法学考察
2.6.1 定量下限、标准曲线、线性范围在国食药监许[2010]455号《化妆品中禁用物质和限用物质检测方法验证技术规范》中,定量下限定义为能够对被测物质准确定量的最低浓度或质量[24]。在保证准确定性的前提下,通过使用空白基质逐级稀释对照品溶液来考察定量下限(LOQ),以信噪比S/N≥10确定73种糖皮质激素的LOQ为0.1~0.3 μg/g(见表2)。
空白样品中添加系列不同质量浓度的73种混合标准溶液,按“1.3”方法处理得到样品提取液,以目标化合物的色谱峰面积(y)对其质量浓度(x,ng/mL)进行线性回归,得到线性方程和相关系数。结果表明,73种目标化合物在各自质量浓度范围内线性关系良好,相关系数均大于0.99(见表2)。
2.6.2 回收率与相对标准偏差选取水剂、膏霜、乳液3类空白基质,按1、2、5倍LOQ进行加标回收实验,每个加标水平平行分析3次。实验数据显示,水剂的平均回收率为88.1 %~118%,相对标准偏差(RSD) 为 0.40%~14%;膏霜的平均回收率为73.3%~119%,RSD为 0.40%~18%;乳液的平均回收率为86.5%~115%,RSD为 0.60%~17%。典型乳液基质的数据见表2。

表2 73种化合物在典型乳液基质中的线性范围、相关系数、回收率、精密度和定量下限Table 2 Linear ranges,correlation coefficients,average recoveries,precisions and limits of quantitation of the 73 compounds in typical emulsion matrices

(续表2)
2.7 实际样品测定
采用本方法对客户委托的7个样品进行了检测,其中2批样品检出倍他米松,含量分别为4.9 μg/g和5.2 μg/g。典型阳性样品的总离子流图及提取离子流图见图3。
3 结 论
本研究建立了通过式高效净化/超高效液相色谱-串联质谱检测化妆品中73种糖皮质激素的分析方法。该方法样品前处理简单,定量准确,灵敏度高,能满足同时测定化妆品中73种糖皮质激素的要求,为有效监管化妆品中非法添加禁用物质提供了技术支撑。
