混合酸溶样氢化物发生–原子荧光光谱法同时测定土壤中的硒和锗
2020-09-26杨小斌张鑫文嘉明陈艾利
杨小斌,张鑫,文嘉明,陈艾利
(核工业二三0 研究所,长沙 410007)
硒和锗是两种非常重要的可维持人体生命活动的微量元素,其中锗有细胞生成促进剂的作用,适当摄入含硒和锗的有机化合物能够提高人体抗肿瘤、消除体内自由基的能力并增强机体免疫力,是帮助人体抵制自然环境污染侵害,防病健身的有效途径,但如果过量摄入硒、锗可能出现中毒反应[1–5]。硒和锗都可以从土壤中通过食物链向人体迁移,人类从食物链中获取适量的硒和锗是最安全有效的可靠途径[6],因此对土壤中硒和锗的含量进行准确的分析具有重要意义。
土壤中硒、锗含量的主要测定方法有光度法、原子吸收(AAS)法、X 射线荧光光谱(XRF)法、电感耦合等离子体发射光谱(ICP–OES)法、电感耦合等离子体质谱(ICP–MS)法[7–11],这几种分析方法均有一些不足之处,如灵敏度低,检出限高,分析时间长,操作繁琐等。其中ICP–MS 法虽然灵敏度高,检出限较低,但用于测定硒时氩气会形成多原子离子(Ar2+),与待测离子质荷比相同而干扰测定,而且ICP–MS 法检测成本较高。绝大部分检测机构在测定土壤中硒和锗的含量时首选氢化物发生原子荧光光谱(HG–AFS)法[12–16]。近年来由于激发光源(特制空心阴极灯)、原子化器、氢化物发生与原子荧光光谱联用分析技术的快速发展,氢化物发生–原子荧光光谱分析技术的应用更为广泛,该方法具有干扰少、灵敏度高、准确度高、检出限低、线性范围宽、操作简便等优点,同时仪器价格低廉也为该方法的推广提供了便利。目前,氢化物发生–原子荧光光谱法可测定各类样品中痕量和超痕量的砷、汞、锑、铋、硒、锗、铅、锡、碲、锌和镉等11 种元素[17–24]。
目前针对土壤中硒和锗含量的测定,国内报道的方法均为硒、锗单元素测定技术,利用氢化物发生–原子荧光光谱法测定土壤中硒和锗的现行国家标准也都是单元素测定法[25–26],标准规定的两种元素的样品前处理方法不同,因为硒的消解体系中需要使用盐酸,且硒在温度高于150℃时会有所损失,而锗在氯离子存在时,86℃以上就会挥发,因此消解体系中若含有氯离子,会导致锗的测定结果偏低[27–29]。
笔者采用硝酸–氢氟酸–高氯酸–浓硫酸混合酸消解体系,在磷酸介质中,硒、锗与硼氢化钾进行氢化物发生反应,进而利用原子荧光光谱法对硒和锗同时测定,消除了硒、锗需要分体系消解且只能单独测定的弊端。
1 实验部分
1.1 主要仪器与试剂
双通道原子荧光光度计:AFS–8800 型,配硒、锗元素空心阴极灯,北京海光仪器有限公司;
石墨电热板:JRY–D350–A 型,湖南金蓉园仪器设备有限公司;
电子分析天平:ML204/02 型,梅特勒–托利多仪器(上海)有限公司;
超存水制备仪:ZWL–PA2–10 型,中沃实验室仪器设备有限公司;
硝酸、硫酸、氢氟酸、高氯酸:均为优级纯,国药集团化学试剂有限公司;
盐酸、磷酸:均为优级纯,成都市科隆化学品有限公司;
三氯化铁晶体(FeCl3·6H2O):分析纯,国药集团化学试剂有限公司;
硼氢化钾、氢氧化钾:均为优级纯,天津市科密欧化学试剂有限公司;
硒、锗标准储备液:质量浓度均为1 000 mg/L,国家有色金属及电子材料分析测试中心;
氩气:纯度为99.999%,长沙赛众特种气体有限公司;
2.0%硼氢化钾–0.5%氢氧化钾溶液:称取20 g 硼氢化钾于预先加有5 g 氢氧化钾的500 mL 超纯水中,用玻璃棒搅拌至溶解,加水稀释至1 000 mL,搅拌均匀,此溶液需现用现配;
土壤成分分析标准物质:编号分别为GBW 07405,GBW 07408,GBW 07429,GBW 07449,GBW 07453,GBW 07457,中国地质科学院地球物理地球化学勘查研究所;
实验所用玻璃器皿均使用20%硝酸溶液浸泡24 h 以上,洗净之后方可使用;
实验用水:超纯水,电阻率为18.25 MΩ·cm。
1.2 仪器工作条件
原子化器高度:8 mm;空心阴极灯电流:硒、锗灯均为80 mA;光电倍增管负高压:270 V;载气流量:300 mL/min;屏蔽气流量:800 mL/min;测量方法:标准曲线法;标准曲线校正方式:单点校正;空白判别值:5;读数方式:峰面积;读数时间14 s;延迟时间:1 s。
1.3 标准曲线绘制
用盐酸溶液(1∶4)将1 000 mg/L 硒标准储备液逐级稀释至1 μg/mL 的硒标准使用液;用磷酸溶液(1∶9)将1 000 mg/L 锗标准储备液逐级稀释至2 μg/mL 的锗标准使用液。取7 只清洗并干燥好的100 mL 容量瓶,分别准确移取0.00,0.10,0.20,0.40,0.80,1.60,2.00 硒 标 准 使 用 液 和0.00,0.25,0.50,1.00,2.00,4.00,5.00 mL 锗标准使用液,依次加入磷酸溶液(1∶1)20 mL,盐酸溶液(1∶1,含铁盐)40 mL,用超纯水定容至100 mL,摇匀,配制成系列混合标准工作溶液。系列混合标准工作溶液中硒的质量溶度依次为0.00,1.00,2.00,4.00,8.00,16.00,20.00 μg/L;锗的质量浓度依次为0.00,5.00,10.00,20.00,40.00,80.00,100.00 μg/L。
仪器开机预热稳定后,测定标准空白,再由低浓度到高浓度依次测定系列混合标准溶液,以标准溶液的质量浓度为自变量、荧光强度为因变量,仪器自动拟合标准曲线线性方程。
1.4 样品前处理方法
称取0.250 0 g 土壤样品(精确至0.1 mg)于25 mL 聚四氟乙烯坩埚中,加几滴水润湿后,依次加入的硫酸溶液(1∶1)1 mL、5~10 mL 硝酸、3 mL 氢氟酸、1 mL 高氯酸,将坩埚置于石墨电热板上恒温加热(电热板温度控制在140℃),加热至刚冒白烟时小心取下;待坩埚自然冷却后,加入10 mL 超纯水,继续温热至盐类溶解,用超纯水将溶液全部转移至25 mL 玻璃比色管中,加入磷酸溶液(1∶1) 5 mL(或浓磷酸2.5 mL )、盐酸溶液(1:1,含铁盐)10 mL,用超纯水稀释至标线,摇匀,溶液中三价铁离子的最终质量浓度为1 mg/mL,放置至澄清。
1.5 样品测定
仪器开机,将原子化器预热30 min 以上,调节至最佳工作条件。以2.0%硼氢化钾溶液–0.5%氢氧化钾溶液作为还原剂,10%磷酸溶液作为载流液,将进样管、还原剂管、载流液管插入相应的试剂溶液中,压紧蠕动泵,导入还原剂及空白介质溶液5~10 min,测定系列混合标准工作溶液。然后在相同的条件下测定样品空白、试样。
2 结果与讨论
2.1 样品消解体系的选择
由于土壤成分相对复杂,不同的消解体系会对测定结果有较大的影响。笔者选用硝酸–氢氟酸–高氯酸–浓硫酸的湿法消解体系。与单一酸消解体系相比,这种混合酸的消解体系不仅试剂用量少,试剂的氧化能力也得到增强;高氯酸的加入可有效破坏土壤中的有机质;同时消解温度控制在140℃可避免因温度过高而导致硒的损失;而采用在样品消解完并冷却之后再加入盐酸,既满足了硒的测量要求,又避免了在温度高于86℃时引入氯离子而造成锗的挥发损失。
2.2 仪器工作条件的选择
2.2.1 原子化器高度
原子化器的高度即火焰的观测高度,是关系到样品是否稳定、测定结果是否准确的先决条件,原子化器高度过小,会使仪器的散射光形成较强的背景信号,信噪比降低,还会产生气相干扰,检出限变高;而原子化器高度过大又会降低仪器的灵敏度和精密度。笔者依据国家标准方法推荐的仪器条件,结合实验室现有的仪器,分别选择8,9,10 mm 3 个不同原子化器高度对硒、锗系列标准溶液的最高浓度点采用双通道同时测定并进行比较,结果列于表1。由表1 可知,当原子化器高度从8 mm 升高至10 mm 时,硒和锗系列标准溶液的最高浓度点荧光强度均有明显下降,而信噪比的增加却不大,因此选择8 mm 作为最佳的原子化器高度。
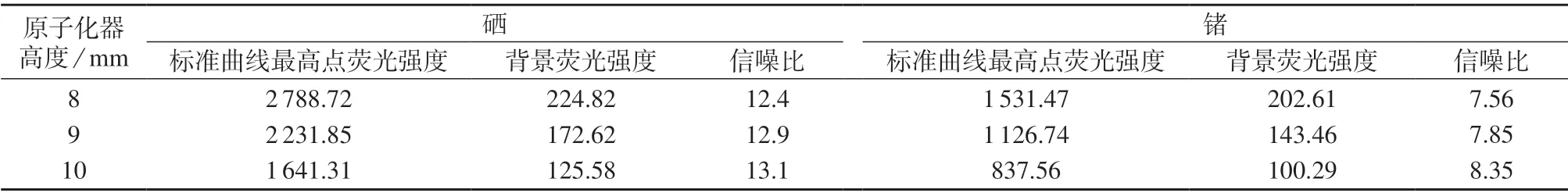
表1 不同原子化器高度试验结果
2.2.2 空心阴极灯灯电流
在一定范围内,待测元素所产生的荧光会随着灯电流的增大而增强,即分析灵敏度提高。但过大的灯电流会产生自吸现象,造成标准曲线弯曲,严重影响重现性,而且会加快空心阴极灯能量的损耗,使灯的使用寿命大幅降低[30],设定灯电流50~100 mA进行试验,结果如图1 所示。
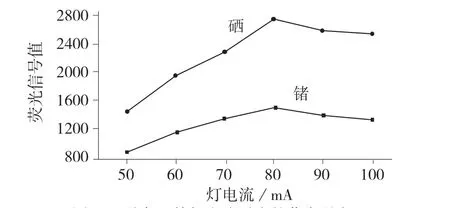
图1 不同硒、锗灯电流对应的荧光强度
由图1 可知,硒灯、锗灯电流为50~80 mA 时,荧光强度不断增强,其中硒的荧光强度增大明显。硒和锗的荧光信号值均在灯电流为80 mA 时达到最高,超过80 mA 之后均呈下降趋势。在满足灵敏度要求的前提下,应尽可能采用较低的灯电流,以便有效降低灯能量的消耗,延长灯的使用寿命,实验最终选择锗、硒灯电流均为80 mA。
2.2.3 光电倍增管负高压
在一定范围内待测元素的荧光强度和仪器测量灵敏度均会随着光电倍增管负高压的增大而提高。负高压过低不能满足灵敏度的要求;而负高压过高则会导致背景噪声增强,同时仪器稳定性变差。依据大量实际样品的测量经验及检测仪器的特性,合适的负高压应控制在系列标准溶液浓度最高点的荧光信号值在1 000~3 000 范围。设定光电倍增管的负高压为220~320 V 进行试验,结果如图2 所示。

图2 不同光电倍增管负高压对应的荧光强度
由图2 可知,硒、锗被激发所产生的荧光强度随着光电倍增管负高压的增大而增强,结合硒和锗元素的特性,选择使锗和硒的标准曲线最高点荧光强度分别达到约1 500 和2 500 的光电倍增管负高压作为最佳负高压。实验选择270 V 的负高压,既能够保证满足信号值、灵敏度、噪声的要求,又有利于延长光电倍增管的使用寿命。
2.2.4 载气流量及屏蔽气流量
适当的载气流量可以保证将氢化物发生反应产生的硒和锗的氢化物迅速导入到原子化器中,并保证稳定的氢–氩火焰。过大的载气流量会稀释待测原子浓度,使荧光强度和分析灵敏度降低;过小的载气流量则会使信号峰拖尾。设定载气流量为200~600 mL/min 进行试验比较,结果如图3 所示。由图3 可知,当载气流量从200 mL/min 增大至300 mL/min 时,硒和锗的荧光强度随之增大;当载气流量大于300 mL/min 时,荧光强度均有所下降,特别是硒的荧光强度下降比较明显。实验确定本方法载气流量为300 mL/min。

图3 不同载气流量对应的荧光强度
屏蔽气对氢–氩火焰起保护作用,可防止空气进入火焰而产生荧光淬灭,稳定火焰状态,保证荧光效率,合适的屏蔽气流量范围应为800~1 100 mL/min。本实验选择屏蔽气流量为800 mL/min。
2.2.5 还原剂的浓度
作为氢化物发生反应的还原剂,硼氢化钾溶液的浓度对方法灵敏度、稳定性、准确度起着决定性作用。硼氢化钾溶液的浓度过高会产生过量的氢,稀释待测原子,甚至引起荧光淬灭,并造成气相和液相干扰;硼氢化钾溶液的浓度过低则氢化物发生反应难以进行。选择硼氢化钾质量浓度为5~30 g/L(其中含5 g/L 氢氧化钾)进行试验,结果如图4 所示。
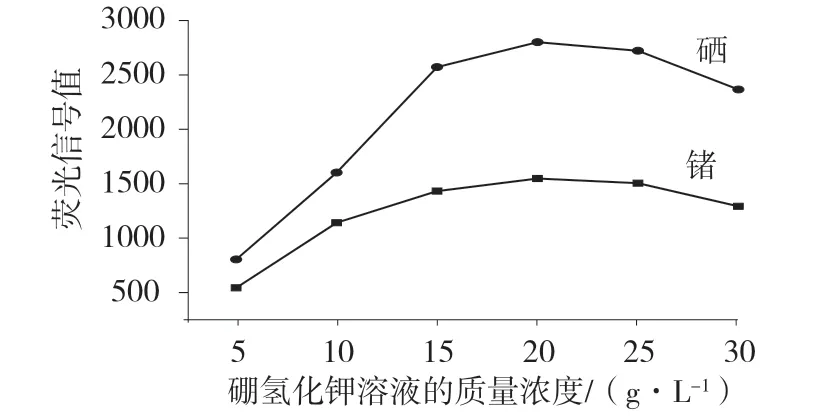
图4 不同浓度的硼氢化钾溶液对应的荧光强度
由图4 可知,当5~15 g/L 的硼氢化钾溶液参与反应时,待测元素产生的荧光强度较低,说明硒和锗没有完全被还原;当硼氢化钾的质量浓度由15 g/L 增大至20 g/L 时,待测元素产生的荧光强度达到最大值;当硼氢化钾的浓度增大到25 g/L 时,待测元素产生的荧光强度呈现下降趋势;当硼氢化钾的浓度增加到30 g/L 时,待测元素产生的荧光强度下降非常明显。可能是因为还原反应产生过量的氢,稀释了GeH4和H2Se,并导致火焰不稳定。在配制硼氢化钾浓液时需加入适量氢氧化钾防止其过快水解,综合考虑荧光强度、灵敏度、稳定性等因素,选定20 g/L 硼氢化钾溶液–5 g/L 氢氧化钾溶液作为还原剂。
2.2.6 载流液的浓度
选择体积分数为2%~20%的磷酸作为载流液,考察其对硒、锗荧光强度的影响,结果如图5 所示。
从图5 可知,随着磷酸浓度增大,荧光强度逐渐增加,当磷酸体积分数为10%时,硒的荧光强度达到峰值,此后随着磷酸体积分数的增加,硒和锗的荧光强度均有所下降,可能是硼氢化钾与磷酸的反应速度太过剧烈,大量的氢以氢气的形式逸出,从而影响了GeH4,H2Se 的生成,降低了方法的灵敏度。因此,在保证足够灵敏度的前提下应尽可能使用较低浓度的磷酸作为载流液。实验选择10%的磷酸作为载流液已能满足要求。
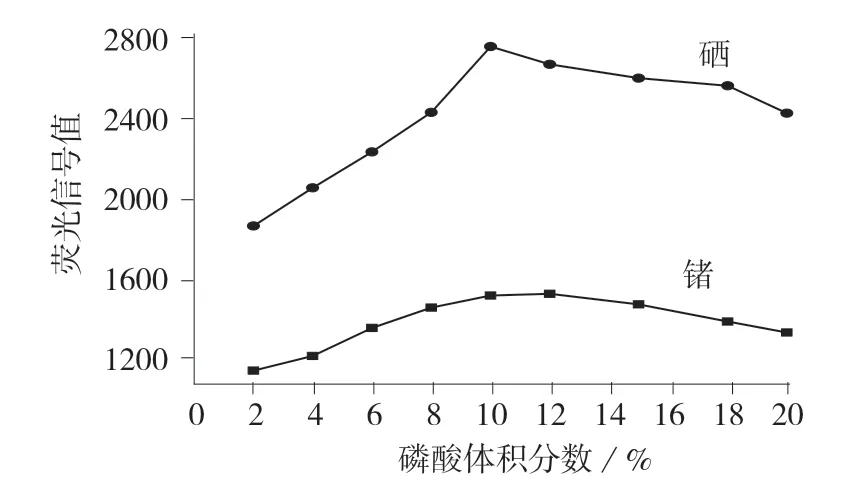
图5 载流液的浓度对锗、硒荧光强度的影响
2.3 线性范围与检出限
在选定的仪器工作条件及测定条件下,对硒和锗的系列混合标准工作溶液进行双通道同时测定,分别以硒、锗的质量浓度(X,μg/L)为自变量、荧光强度(Y)为因变量进行线性拟合,得硒、锗的线性方程分别为Y=139.98X+1.27,Y=15.44X+9.91,相关系数(r2)分别为1.000,0.999 3。硒、锗的线性范围分别为0~20,0~100 μg/L。平行进行7 次空白试验,按照土壤称样量为0.25 g,定容体积为25 mL,计算硒、锗测定结果的标准偏差s分别为0.003,0.006 mg/kg,按MDL=stn–1,0.99计 算 方 法 检 出 限,当n=7 时,t值取3.143[31],得硒、锗的检出限分别为0.010,0.020 mg/kg。
2.4 标准样品测定结果
按照1.4 的样品前处理方法对编号分别为GBW 07405,GBW 07408,GBW 07429,GBW 07449,GBW 07453,GBW 07457 的土壤国家标准物质进行消解,然后双通道同时测定硒、锗的含量,平行测定6 次,与标准物质硒、锗含量标示值进行比较,计算相对误差,并与单道测定结果进行比较,结果见表2。
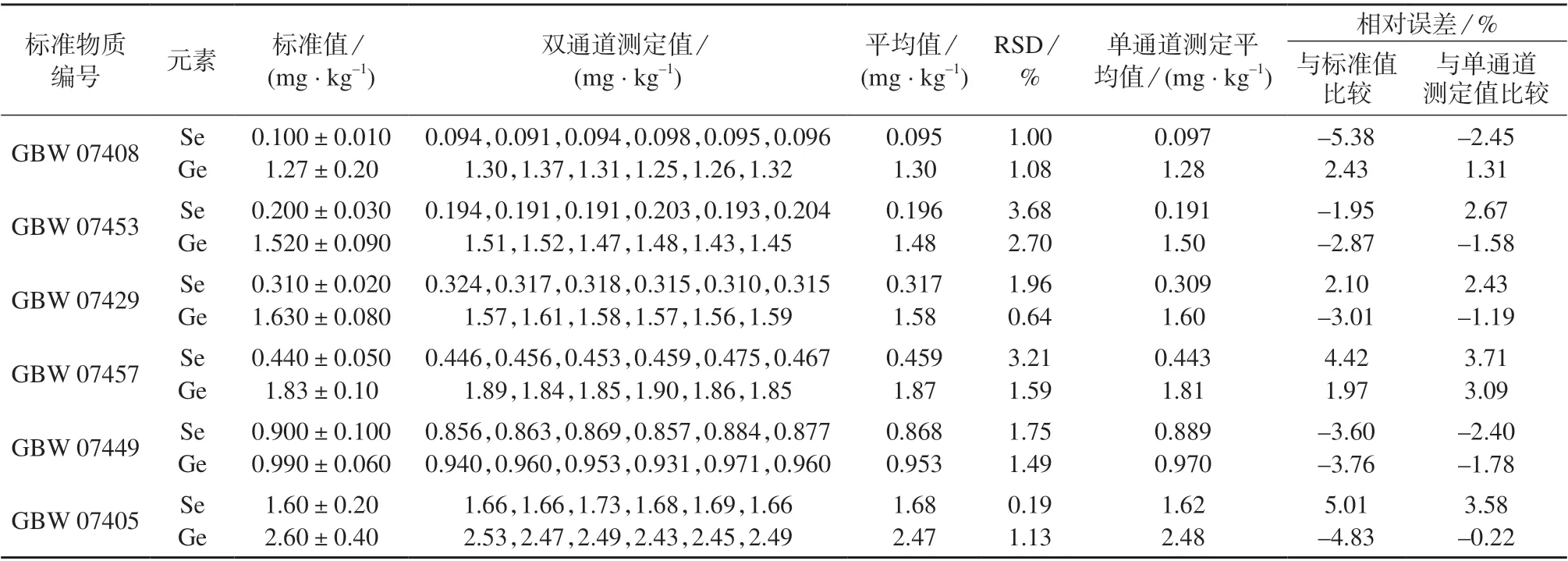
表2 准确度与精密度试验结果
由表2 可知,不同含量的标准物质硒、锗测定结果均在标准物质标准值范围内,相对误差分别为–5.38%~5.01%,–4.83%~2.43%,与单通道测定结果相对误差分别为–2.45%~3.71%,–1.78%~3.09%。双通道同时测定结果的相对标准偏差分别为0.19%~3.68%,0.64%~2.70%(n=6)。依据土壤环境监测技术规范,该方法同时测定土壤中硒和锗时,准确度和精密度均能满足质量控制要求[32]。
3 结语
针对大批量土壤样品中硒和锗的测定需求,选定硫酸–硝酸–氢氟酸–高氯酸组成的消解体系,建立了双通道原子荧光光谱法同时测定硒和锗含量的方法。该方法检出限低,灵敏度高,精密度好,准确度高,成本低,消除了锗和硒需要分体系消解、一次只能单通道测定一种元素的弊端,具有良好的应用前景。
