高效液相色谱–串联质谱法测定水中唑啉草酯和炔草酯
2020-09-26杨浪陈恺王波娜廖朝选魏杰谢源
杨浪,陈恺,王波娜,廖朝选,魏杰,谢源
(1.贵州省分析测试研究院,贵阳 550014; 2.贵州健安德科技有限公司,贵阳 550014)
唑啉草酯(Pinoxaden)是一种新苯基吡唑啉类除草剂,具有杀草谱较宽、除草效果好、对环境友好的特点,而且杂草对其较难产生抗性。唑啉草酯与其它传统除草剂无交互抗性,是防除麦田禾本科杂草药剂中安全性最高的农药之一[1]。炔草酯(Clodinafop-propargyl)是芳氧苯氧基丙酸类(AOPP)专门防除禾本科杂草的除草剂,具有吸收快、起效快、耐雨水冲刷、低温下稳定的特点[2]。两种农药对禾本科植物都有优异的防治效果,同时各具特点,混配后对小麦田杂草预防效果良好[3],当前在我国已有应用。农药应用于田间后,可能因随雨水径流、土壤淋溶等进入自然水体,影响水中生物生长,从而造成水生态系统风险,分析两种农药在水中的残留量,对评估农药产生的生态风险非常必要[4–5]。
关于两种农药的分析,俞霖洁等[6]、谢俊婷等[7]分别使用乙腈–水、乙腈–0.01%磷酸溶液建立了同时测定唑啉草酯和炔草酯含量的高效液相色谱分析方法;潘静等[8]、杨闻翰等[9]采用高效液相色谱法分别测定了炔草酯和唑啉草酯,郭玉香[10]采用电子捕获检测器气相色谱法测定了麦田土壤中的炔草酯。农药在农作物及土壤中的残留量通常较低,高效液相色谱分析法的检出限较难满足分析要求,液相色谱–串联质谱法是常用于痕量农药残留的分析手段[11–12],具有更高的灵敏度。井静等[13]利用液相色谱–串联质谱联用法测定了小麦和麦秆中的炔草酯;徐潇颖等[14]利用液相色谱–串联质谱联用法进行了炔草酯在蔬菜中的残留分析。关于水中唑啉草酯和炔草酯的分析目前未见报道。
笔者以甲醇–0.1%甲酸水溶液作为流动相,建立了同时测定水中唑啉草酯和炔草酯含量的液相色谱–串联质谱分析方法,可用于唑啉草酯和炔草酯相关产品的水生毒性试验浓度测定与水中农药残留分析。
1 实验部分
1.1 主要仪器与试剂
液相色谱–串联质谱联用仪:Agilent 1260–G6465B 型,美国安捷伦科技有限公司;
电子天平:XSE105DU 型,梅特勒–托利多仪器(中国)有限公司;
唑啉草酯标准品:纯度为99.19%,国家农药质量监督检验中心;
炔草酯标准品:纯度为99.19%,德国Dr. Ehrenstorfer 公司;
甲醇:色谱纯,批次为SHBK6874,德国默克公司;
10%唑啉草酯·炔草酯可分散油悬浮剂:唑啉草酯、炔草酯的质量分数均为5%,贵州省分析测试研究院;
地表水样品:曝气生态水,充氧并经活性炭脱氯48 h,pH=7.8;
实验用水:去离子水,自制。
1.2 实验方法
1.2.1 混合标准溶液的制备
准确称取0.010 1 g 唑啉草酯和0.010 2 g 炔草酯于称量瓶中,用色谱纯甲醇少量多次溶解后,将其定量转入100 mL 容量瓶中,定容至标线,摇匀,得到两种农药质量浓度均为100 mg/L 的混合标准母液。然后分别量取不同体积的母液,用甲醇稀释配制成质量浓度分别为0.10,0.20,0.50,1.0,2.50,5.0 μg/L 的系列混合标准溶液。
1.2.2 试样溶液的制备
准确称取0.010 2 g 10%唑啉草酯· 炔草酯可分散油悬浮剂于称量瓶中,然后用曝气生态水少量多次转入1 L 烧杯中,制备得1.02 mg/L 的试样溶液,稀释至500 倍体积,过0.45 μm 滤膜。
1.2.3 加标样品的制备
取曝气生态水作为基质样品,然后分别向1 L基质样品中添加0.05,0.50 μg 炔草酯和唑啉草酯作为基质加标样品。
1.3 仪器工作条件
1.3.1 高效液相色谱
色谱柱:Eclipse Plus-C18柱(50 mm×2.1 mm,1.8 µm,SN:USDAY55083,美国安捷伦科技有限公司);进样体积:10 μL;流动相:甲醇–0.1%甲酸水溶液(90∶10),流量为0.3 mL/min;柱温:30℃。
1.3.2 质谱
离子源:AJS ESI 源,正离子模式;干燥气:N2;干燥气温度:300℃;干燥气流量:7 L/min;鞘气:N2;鞘气温度:250℃;鞘气流量:11 L/min;雾化气压力:241.3 kPa;喷嘴电压:1 000 V;毛细管电压:4 000 V;监测模式:质谱多反应监测(MRM),MRM 离子采集参数见表1。
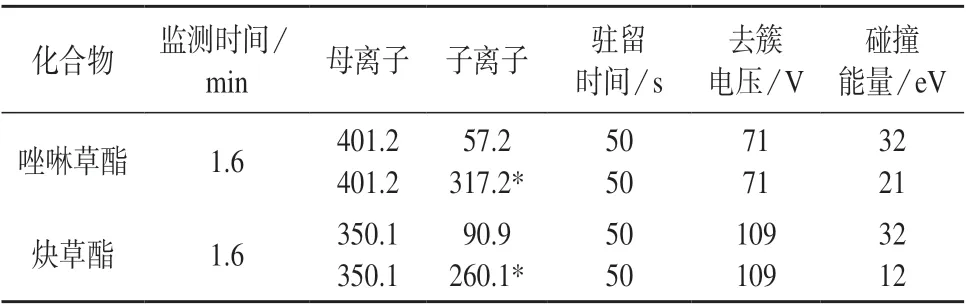
表1 MRM 离子采集参数
1.4 样品测定
在1.3 仪器工作条件下,待仪器基线稳定,且相邻2 次标准样品溶液的色谱峰面积相对变化小于1.5%时,按混合标准溶液–试样溶液–试样溶液–混合标准溶液的次序进行测定。
1.5 结果计算
将试样溶液两次测定结果与前后各一次的标准样品中唑啉草酯和炔草酯色谱峰面积分别取平均值,利用式(1)计算唑啉草酯和炔草酯的含量:

式中:w——唑啉草酯或炔草酯的质量分数;
A1,A2——标准溶液、试样溶液中唑啉草酯或 炔草酯色谱峰面积的平均值;
m1,m2——标准样品、试样溶液中唑啉草酯或 炔草酯的质量,g;
P——标准样品中唑啉草酯或炔草酯的质量 分数。
2 结果与讨论
2.1 流动相的选择
用液相色谱–串联质谱法测定炔草酯或唑啉草酯时,井婧等[12]使用乙腈–水作为流动相,韩何丹等[13]采用乙腈–0.1%甲酸水作为流动相。唑啉草酯和炔草酯均溶解于乙腈,也溶解于甲醇,使用甲醇作为流动相时分析时间较乙腈短,而两个组分的色谱峰面积、峰形未见明显差异,因此选择甲醇–水体系作为流动相。在该体系中,甲醇比例较低时,色谱峰拖尾严重,逐步增加甲醇比例,峰形明显改善;而且在水相中添加0.1%甲酸后可增强目标物的质子化,使色谱峰形更加理想,灵敏度更高[15],最终确定最佳流动相为甲醇–0.1%甲酸水溶液(90∶10)。
2.2 质谱条件优化
以甲醇–0.1%甲酸水溶液(90∶10)为流动相,选择10.0 μg/L 的唑啉草酯和炔草酯混合标准工作溶液注入离子源中,在正负离子监测模式下对目标物进行全扫描以确定目标物母离子。根据不同传输电压下母离子的响应值确定最佳传输电压,并根据响应值最高的碎片离子确定定量和定性子离子,然后确定碰撞能量并对其它参数进行优化,优化后的多反应监测(MRM)条件见表1。
2.3 方法选择性
分别测定空白基质样品、质量浓度均为1.0 μg/L 的唑啉草酯、炔草酯混合标准溶液,在仪器最高灵敏度下空白样品无可见干扰,唑啉草酯标准样品的保留时间为0.79 min,炔草酯标准样品的保留时间为0.76 min。
2.4 线性方程与检出限、定量限
在1.3 仪器工作条件下,分别测定唑啉草酯、炔草酯系列混合标准溶液,以唑啉草酯、炔草酯的质量浓度(x,μg/L)为自变量,对应的色谱峰面积(y)为因变量进行线性拟合,得到唑啉草酯、炔草酯的线性方程。结果表明:唑啉草酯、炔草酯的质量浓度在0.10~5.0 μg/L 范围内与色谱峰面积具有良好的线性关系,回归方程分别为y=14 858.98x+154.31,y=2 042.03x+15.44;相关系数(r)分别为0.999 5,0.999 7。以信噪比3 确定唑啉草酯和炔草酯的检出限分别为1.8×10–3,3.1×10–3μg/L;以信噪比10确定唑啉草酯和炔草酯的定量限分别为6.0×10–3,1.0×10–2μg/L。
2.5 加标回收试验
取曝气生态水,进行两个浓度水平的基质加标回收试验,每个加标水平平行试验6 次。分别用本法测定样品本底及加标样品中唑啉草酯和炔草酯的含量,加标样品质谱多反应监测色谱图如图1 所示。计算唑啉草酯和炔草酯的回收率和相对标准偏差,相关数据列于表2。由表2 可知,在0.10,0.50 μg/L 两个加标水平下,唑啉草酯、炔草酯的回收率分别为93.0%~106.4%,93.0%~108.6%,相对标准偏差分别为3.90%,4.46%,表明本方法的精密度、准确度良好。

图1 地表水加标样品质谱多反应监测色谱图

表2 加标回收试验结果
3 结语
利用高效液相色谱–串联质谱联用法同时测定地表水中的唑啉草酯、炔草酯残留,该方法的准确度与精密度良好,操作简单、快速,有推广应用价值。
