超临界流体色谱—串联质谱分离和测定烟草中磷胺和速灭磷顺反异构体
2020-09-25杨飞孟红明崔浩哲范多青王颖刘珊珊范子彦唐纲岭赵蔚
杨飞,孟红明,崔浩哲,范多青,王颖,刘珊珊,范子彦,唐纲岭,赵蔚
1 国家烟草质量监督检验中心,郑州高新技术产业开发区翠竹街6号 450001;
2 云南中烟工业有限责任公司,昆明市五华区红锦路181号 650023;
3 苏州科技大学化学生物与材料工程学院,苏州市高新区科锐路1号 215009
顺反异构是一种重要的立体异构现象。化合物分子中由于具有限制旋转的因素,使各个基团在空间的排列方式不同而出现顺反异构[1],这些异构体的性质不完全相同。化合物分子中有双键或是环状结构经常会有这种现象。其中双键的顺反异构又以C=C 双键最为常见,此外还有C=N 双键、N=N 双键、C=S 双键等。构型不同的化合物,生物活性差别较大,有时可能达到几十甚至几百倍[2]。例如,啶菌恶唑Z、E 异构体虽然杀菌谱均较广,有较好的抑菌活性,但对部分真菌活性存在差异[3]。灭多威E 体杀虫活性也远小于Z 体,对巢修尾蚜的室内药效测定表明,E 体仅为Z 体的1/40;对家蝇成虫的药效测定也表明E 体活性只有Z 体的1/10,且Z体较E 体稳定[4]。又如辛硫磷Z 体对蚊幼虫的杀虫活性明显优于E 体,Z 体的杀虫活性比E 体高约3倍[5]。
磷胺又名大灭虫,在中性和弱酸性溶液中较稳定,在碱性液中会迅速分解。磷胺对人、畜毒性大,属高毒农药。磷胺对烟草上的害虫和螨类具有强烈的触杀和胃毒作用。磷胺为广谱性有机磷杀虫剂,可防治刺吸式口器和咀嚼式口器的多种害虫。磷胺分子结构中含有C=C,具有一对顺反异构体(图1),其中顺式异构体具有较强活性[6]。速灭磷又名磷君,在碱性条件下分解较快。速灭磷为触杀性,内吸性杀虫、杀螨剂。速灭磷分子结构中含有C=C,具有一对顺反异构体(图1),在植物体内顺式异构体比反式异构体代谢速度快,其顺反异构体毒性也不尽相同,顺式异构体比反式异构体毒性大20 倍左右[7]。

图1 磷胺和速灭磷的化学结构Fig. 1 Chemical structures of phosphamidon and mevinphos
在国际烟草科学研究合作中心(Cooperation Centre for Scientific Research Relative to Tobacco,CORESTA)农用化学品咨询委员会(Agro-Chemical Advisory Committee,ACAC) 制 定 的106 种 农 用化学品指导性残留限量表(Agrochemical Guidance Residue Levels,GRL)中,速灭磷和磷胺的限量均是按各异构体总和计,速灭磷(E-速灭磷和Z-速灭磷之和)和磷胺(E-磷胺和Z-磷胺之和)的残留限量分别为0.04 mg/kg 和0.05 mg/kg[8]。
目前未见关于磷胺和速灭磷顺反异构体拆分和测定的报道。顺反异构体的拆分主要采用液相色谱(HPLC)[9-12],该方法需要消耗大量有毒有害的有机溶剂,而且分析时间长。如李大婧等用HPLC 分离和测定了叶黄素的顺反异构体,分析时间长达30 分钟,且消耗较多的有机溶剂[10]。近几年来,作为一种新的分析技术,超临界流体色谱(SFC)引起了研究人员越来越多的关注[13-14]。SFC 主要以超临界CO2和少量的有机溶剂为流动相,与HPLC 相比,分析速度快、时间短、分离效率高。同时,SFC 的有机溶剂消耗量低于HPLC,更环保。这些优点使得SFC 已广泛应用于不同基质中手性化合物的分析,测定和制备分离[15-23]。
本文通过超临界流体色谱串联质谱(SFC-MS/MS)首次研发了一种环境友好,灵敏,快速的顺反异构体分离和测定的方法,用于选择性拆分和测定烟草中的磷胺和速灭磷顺反异构体。而且,SFC 和MS/MS 的组合可以进一步提高方法的灵敏度。
1 材料与方法
1.1 试剂和材料
20 个烤烟实际样品全部来自2018 年度国家烟草专卖局农残普查分析样品。所有样品烘干后粉碎,并过2 mm(10 目)筛后贮存于棕色玻璃瓶中,4℃下保存。
E-磷胺标准品(CAS:297-99-4)、Z-磷胺标准品(CAS:23783-98-4),E-速灭磷标准品(CAS:298-01-1)、Z-速灭磷标准品(CAS:338-45-4)(纯度>98.0%,天津阿尔塔科技有限公司)。甲醇、乙醇、乙腈、异丙醇和甲酸(色谱纯,德国Merck 公司);无水硫酸镁(MgSO4)、氯化钠(NaCl)、N-丙基乙二胺(PSA)(分析纯,上海安普实验科技股份有限公司);水为超纯水;高纯氮(纯度> 99.99%,郑州源正科技);高纯氩(纯度>99.99%,郑州源正科技)。
ACQUITY 超临界流体色谱仪、ACQUITY TQD四极杆串联质谱仪(配电喷雾电离源,美国Waters公司);Acquity UPC2Trefoil AMY1、Acquity UPC2Trefoil CEL1 和Acquity UPC2Trefoil CEL2 柱(100 mm×2.1 mm,1.7 μm)( 美 国Waters 公 司);Chiralpak IG-3 柱(100 mm×3.0 mm,3.0 μm)(日本Daicel 公司);VX200 涡旋振荡仪(美国Labnet公司);SG3-30K 高速离心机(德国sigma 公司);Milli-Q 超纯水系统(美国Millipore 公司);CP224S电子天平(感量:0.0001 g,德国Sartorius 公司)。
1.2 方法
1.2.1 样品前处理
样品前处理过程采用本文作者之前的研究[24]。称取2 g 样品(精确至0.01 g)于50 mL 具盖离心管中,加入10 mL 超纯水和10 mL 乙腈,然后以2000 rpm 的速率涡旋振荡2 min。向离心管中加入4 g 无水硫酸镁和1 g 氯化钠、1 g 柠檬酸钠和0.5 g 柠檬酸二氢钠,继续以2000 rpm 的速率涡旋混合2 min。移取1 mL 上清液于1.5 mL 离心管中,加入150 mg 无水硫酸镁及25 mg PSA 吸附剂,于漩涡混合振荡仪上以2000 rpm 的速率振荡2 min,以6000 rpm 的速率离心5 min。吸取上清液经0.22 μm 有机相滤膜过滤,取200 μL 的滤液加入800 μL 的乙腈,混匀,进SFC-MS / MS 检测。
1.2.2 基质匹配标准工作溶液的配制
分别称取10 mg 的E-磷胺,Z-磷胺,E-速灭磷,Z-速灭磷标准品于100 mL 容量瓶中,用乙腈溶解并定容至刻度,制得E-磷胺、Z-磷胺、E-速灭磷和Z-速灭磷的质量浓度均为100 μg/mL 的一级混合标准储备液。
移取1.0 mL 一级混合标准储备液于100 mL 容量瓶中,用乙腈定容,得到E-磷胺、Z-磷胺、E-速灭磷和Z-速灭磷的质量浓度均为1.0 μg/mL 二级混合标准储备液。
分别移取二级混合标准储备液500 μL、200 μL、100 μL、50 μL、20 μL 至5 个10 mL 容量瓶中,用乙腈定容,制得不同质量浓度的标准工作溶液。
分别移取0.2 mL 标准工作溶液和0.2 mL 空白样品提取液,混合后用乙腈稀释至1 mL。各基质匹配标准工作溶液中各异构体的质量浓度分别为10.0 ng/mL、4.0 ng/mL、2.0 ng/mL、1.0 ng/mL 及0.4 ng/mL。所有溶液避光储存在-20℃的冰箱中。
1.2.3 加标样品的制备
在最优化的试验条件下,通过对实际样品的分析,选取1 个烟叶样品作为空白样品。添加不同含量(20 μL、100 μL、400 μL)的二级混合标准储备液到2.0 g空白样品中,获得三个不同浓度的空白加标样品,加标样品中各异构体的含量为0.01 mg/kg、0.05 mg/kg、0.2 mg/kg。然后在样品中加入10 mL 甲醇并用涡旋振荡混匀10 分钟。之后,放置于通风橱中在室温下让溶剂蒸发。加标样品按照1.2.1 节方法处理,每个浓度水平重复测定5 次,连续测定3 天,采用基质匹配标准工作曲线定量分析。
1.3 检测条件
色 谱 柱:Acquity UPC2Trefoil AMY1 柱(150 mm×3 mm,2.5 μm);背压:13.79 MPa;柱温:35℃;样品室温度:10℃;补偿溶剂:0.1%甲酸的甲醇溶液,流速0.2 mL/min;进样量:2 μL;流动相流速:2.0 mL/min;流动相:A 相为CO2,B 相为乙醇;梯度洗脱程序:初始至第2 分钟CO2和乙醇的体积比由99% : 1%变成92% : 8%;第2 分钟到第3.5 分钟CO2和乙醇的体积比从92%:8%变成88% : 12%;第3.5 分中到第4 分钟CO2和乙醇的体积比从88% : 12%变成99% : 1%;第4 分钟到第5分钟CO2和乙醇的体积比为99% : 1 %。质谱扫描方式:正离子扫描;离子源:电喷雾电离源(ESI);离子源温度:150℃;脱溶剂气温度:350℃;脱溶剂气流量:650 L/h;锥空气流量:60 L/h;停留时间:100 ms;毛细管电压:3.5 KV;采集模式:多反应监测(MRM);质谱参数见表1。
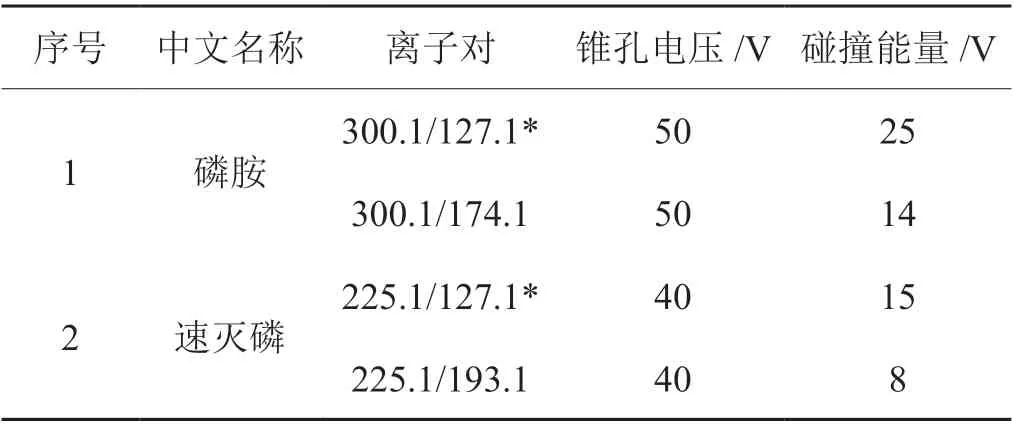
表1 磷胺和速灭磷的质谱参数Tab. 1 MS parameters for phosphamidon and mevinphos
1.4 数据分析
数据采集采用MassLynxV4.1 软件。平均值、回收率和相对标准偏差的计算采用Microsoft Excel 2007版软件。
2 结果和讨论
2.1 手性分离条件的优化
2.1.1 手性柱的选择
分别考察了四种不同的手性柱(Acquity UPC2Trefoil CEL1,Acquity UPC2Trefoil CEL2,Acquity UPC2Trefoil AMY1,Chiralcel IG-3)对磷胺和速灭磷顺反异构体的分离情况[25,26]。Acquity UPC2Trefoil CEL1 和Acquity UPC2Trefoil CEL2 均为改性的多聚糖型固定相,其中Acquity UPC2 Trefoil CEL1 硅胶表面涂敷有纤维素-三(3,5-二甲基苯基氨基甲酸酯),Acquity UPC2Trefoil CEL2 硅胶表面涂敷有纤维素-三(3-氯-4-甲基苯基氨基甲酸酯),Chiralpak IG-3硅胶表面共价键合有直链淀粉-三(3-氯-5-甲基苯基氨基甲酸酯),Acquity UPC2Trefoil AMY1 硅胶表面共价键合有直链淀粉-三(3,5-二甲基苯基氨基甲酸酯)。
当 使 用Acquity UPC2Trefoil CEL1 和Acquity UPC2Trefoil CEL2 时,不能实现磷胺和速灭磷顺反异构体的完全分离。Acquity UPC2Trefoil AMY1,Chiralcel IG-3 均可实现对映体的分离,但是使用Acquity UPC2Trefoil AMY1 时,异构体之间的分离度较好。从分离及色谱峰峰形综合考虑,选用Acquity UPC2Trefoil AMY1 柱对磷胺和速灭磷异构体进行分离(图2)。
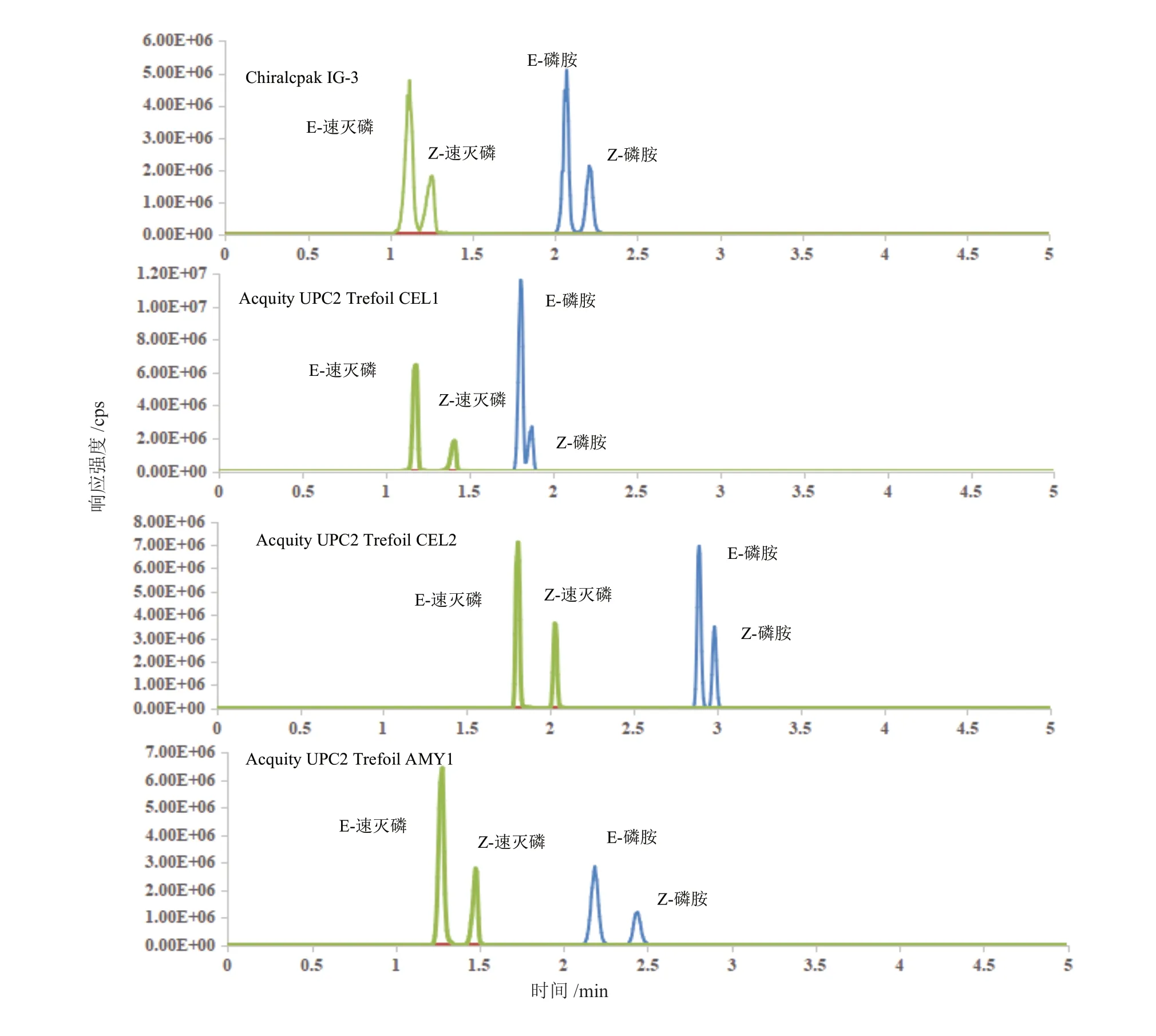
图2 在四种不同色谱柱上磷胺和速灭磷顺反异构体的SFC-MS/MS 色谱图Fig. 2 Typical SFC-MS/MS chromatograms of isomers of phosphamidon and mevinphos on four columns
2.1.2 改性剂的影响
超临界流体色谱以非极性的超临界CO2流体为流动相,其对极性化合物的溶解性能和洗脱能力较弱,为增强流动相对分析物的溶解性能和洗脱能力,达到更好的分离效果和改善峰形,往往在超临界二氧化碳流体中加入少量的极性有机溶剂(改性剂)[27,28]。在Acquity UPC2Trefoil AMY1 手性柱上,不添加任何改性剂即以纯超临界二氧化碳为流动相条件下(其他色谱条件为温度35℃,系统背压13.79 Mpa,流动相流速2 mL/min),磷胺和速灭磷均不能被洗脱(洗脱时间大于30 min)。这是由于这几种物质分子极性大,与手性柱产生较强的相互作用力使得洗脱困难造成的。如图3 所示,本文对三种有机改性剂(甲醇、乙醇和异丙醇)对磷胺和速灭磷顺反异构体在Acquity UPC2Trefoil AMY1 柱上的分离情况进行了考察。如图3 所示,使用异丙醇作为改性剂后面一组峰的分离度较差,使用甲醇和乙醇作为改性剂均可以实现磷胺和速灭磷异构体的基线分离,但乙醇更为环保。实验中还考察了甲酸、乙酸和甲酸铵对磷胺和速灭磷异构体分离的影响,结果表明,加入甲酸、乙酸等并没有改善异构体之间的分离度和响应强度。因此,最终选择乙醇作为流动相改性剂。
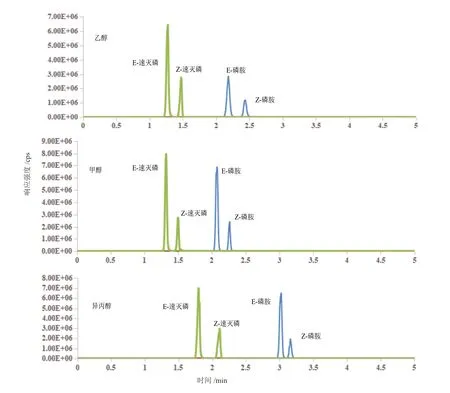
图3 改性剂对磷胺和速灭磷顺反异构体分离情况的影响Fig. 3 The effect of modifier solvent on the separation of isomers of phosphamidon and mevinphos
2.1.3 系统背压的影响
系统背压的变化会改变流动相的密度,从而影响对目标化合物的洗脱能力和选择性。随着系统背压的升高,超临界CO2流体密度随着增加,对分析物的溶解和洗脱能力增强,目标物的保留时间随着减小[29]。分别考察了不同的背压(11.03 Mpa、12.41 Mpa、13.79 Mpa 和15.17 Mpa)对磷胺和速灭磷异构体的分离情况。如图4 所示,随着背压的增加,保留时间减小,但是背压对异构体的分离效率没有显著影响,磷胺与速灭磷异构体的分离度几乎不变。但是当背压为13.79 MPa 时对映体的响应强度最高,而且较高的压力会缩短手性柱的使用寿命。因此,选择13.79 Mpa 的系统背压作为推荐值。

图4 背压对磷胺和速灭磷顺反异构体分离情况的影响Fig. 4 The effect of ABPR on the separation of isomers of phosphamidon and mevinphos
2.1.4 柱温的影响
一般情况下,在液相色谱中,随着温度的升高,目标化合物在固定相上的吸附/脱附会加快,从而加快样品的分离。而在SFC 中,随着柱温的升高,超临界CO2流体的密度和粘度降低,从而减慢了目标化合物在固定相上的吸附/脱附过程,导致保留时间延长[30,31]。在背压13.79 MPa、流速2 mL/min 的条件下,研究了不同温度(35℃、40℃、45℃、50℃)下磷胺和速灭磷顺反异构体的分离效果。如图5 所示,随着温度的升高,保留时间增加,但是异构体之间的分离度几乎不变。因此选择35℃作为最优色谱柱温度。
图5 柱温对磷胺和速灭磷顺反异构体分离情况的影响Fig. 5 The effect of column temperature on the separation of isomers of phosphamidon and mevinphos
2.1.5 磷胺和速灭磷顺反异构体的洗脱顺序
在优化的色谱条件下分别将E-磷胺和E-速灭磷标准溶液注入SFC-MS/MS 中来鉴定每种异构体的洗脱顺序。磷胺和速灭磷顺反异构体在手性柱(Acquity UPC2Trefoil AMY1)上按以下顺序洗脱:E-速灭磷(1.33 min)、Z-速灭磷(1.48 min)、E-磷胺(2.37 min)、Z-磷胺(2.48 min)。在最优化的分离条件下,烟草实际加标样品的选择离子色谱图如图6 所示。

图6 烟草实际加标样品的色谱图(0.2 mg/kg)Fig. 6 Chromatogram of actual spiked samples of tobacco (0.2 mg/kg)
2.2 方法评价
2.2.1 基质效应[32]
以各个异构体在烟草基质中的线性方程斜率与其在乙腈中的线性方程斜率的比值来考察基质效应,当比值在0.9~1.1 之间时,基质效应不明显,当比值大于1.1 时为基质增强效应,小于0.9 则为基质抑制效应。结果(表2)表明,各个异构体的基质效应在0.10~0.75 之间,说明在烟草基质中存在显著的基质抑制效应。因此,烟草样品中所有异构体的测定均使用基质匹配标准溶液进行定量。
对样品基质进行稀释可以降低基质效应。本文考察了不同稀释倍数下烤烟样品中各对映体的基质效应,由表2 可知,随着稀释倍数的增加,分析物的相对响应强度(各样品的响应强度与标样的响应强度之比)也随着增大,表明稀释有助于减小基质效应。但是稀释倍数过高,分析物浓度则降低,影响方法灵敏度。因此,选用乙腈将其稀释5 倍进样。
2.2.2 标准曲线、检出限和定量限
选用空白烟叶试样按1.2 节实验步骤提取净化后,添加不同级别的混合标准工作溶液配制成基质匹配标准工作溶液,进行SFC-MS/MS 分析。通过各农药异构体的峰面积对其浓度进行回归分析,得到标准曲线、相关系数。标准曲线的形式为y=Ax+B,其中A 和B 分别表示为斜率和截距。对于每种异构体,在各自的浓度范围内分析纯溶剂标准工作曲线和不同基质匹配的标准工作曲线,以确定该方法的线性关系。按信噪比(S/N)=3 确定方法检出限,信噪比(S/N)=10 确定方法定量限。结果表明(表2),不同基质中各异构体线性关系良好(相关系数R2≥0.9922),LOD 和LOQ 范围分别为0.6 μg/kg~1.7 μg/kg 和2.0 μg/kg~5.5 μg/kg,可以满足定量分析的需要。

表2 各异构体的线性方程、基质效应、检出限和定量限Tab. 2 Regression equation, matrix effect, LODs, and LOQs for the isomers of phosphamidon and mevinphos
2.2.3 方法的回收率及精密度
将空白烟叶样品分成3 份,分别添加0.02 mL、0.1 mL 和0.4 mL 的混合标准溶液,每个浓度的样品每天重复测定5 次,重复试验3 天,按照1.2.1 节的方法进行前处理。通过空白样品加标回收实验来测试该方法的准确度和精确度,其精确度由重复性实验确定,并以RSD 表示。通过比较同一天内加标样品的回收率的标准偏差来测量日内精密度。通过分析三个不同日期的加标样品来确定日间精确度。结果(表3)表明,在所有浓度水平下的加标回收率为84.7%~96.7%,日内精密度(RSD)为2.0%~4.2%,日间精密度(RSD)为2.4%~4.3%。说明在所有浓度水平下均获得令人满意的回收率,且精密度良好,该方法适用于烟草中磷胺和速灭磷异构体的分离和测定。
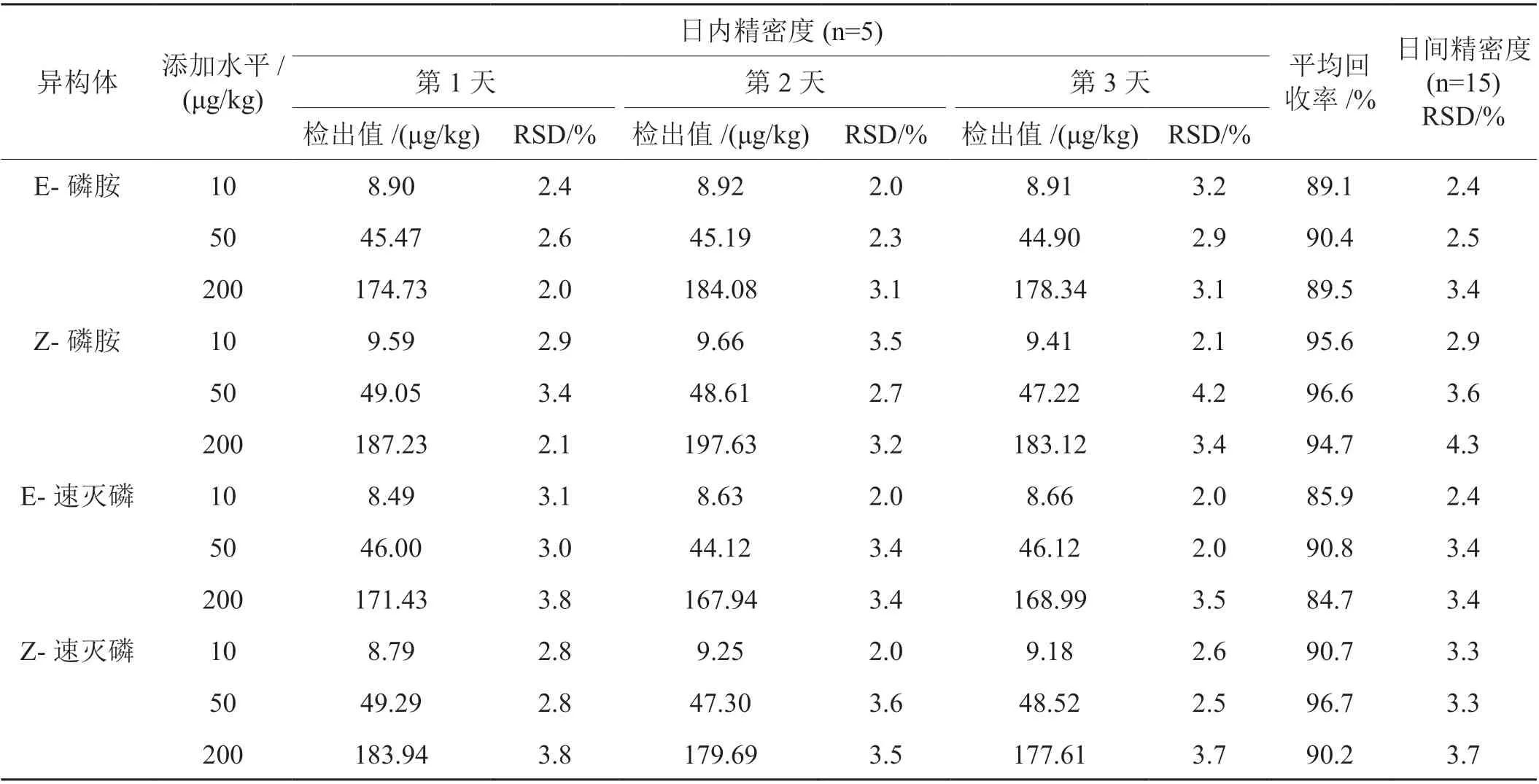
表3 方法的精密度和准确度Tab. 3 Accuracy and precision of the proposed method
采用本方法对20 个烤烟样品进行了测定,所有样品均未检出磷胺和速灭磷的异构体。
3 结论
本文采用超临界流体色谱与串联质谱联用建立了分离和测定磷胺和速灭磷顺反异构体的方法。在最优化的实验条件下,磷胺和速灭磷顺反异构体的回收率在84.7%~96.7%之间,日内精密度为2.0%~4.2%,日间精密度为2.4%~4.3%。该方法以CO2和乙醇作为流动相,绿色环保、简单快速、灵敏度高、选择性强、结果准确,在5 min 内即可实现磷胺和速灭磷顺反异构体的基线分离,在实际样品的批量检测中有巨大的优势。
