唾液酸检测方法研究进展*
2020-09-24焦淑玲朱美旗高文运
杨 阳,焦淑玲,朱美旗,高文运
(1.西安医学院 药学院,药物研究所,陕西 西安710021;2.西北大学 生命科学学院 国家微检测系统工程研究中心,陕西 西安 710069)
唾液酸(SAs)是以 9 个碳为母链的 α-酮酸[1],由Gunnar Blix 等人由颌下腺粘蛋白中分离出来,是一种天然的碳水化合物。目前,唾液酸衍生物的类型已超过50 种,主要包括3 种基本形式:N-乙酰神经氨酸(Neu5Ac)、N-羟基神经氨酸(Neu5Gc)和 3-脱氧-D-甘油-D-半乳壬酮糖(KDN)[2]。唾液酸带负电荷,广泛存在于哺乳动物细胞表面,在细胞之间的信息传递过程中发挥着非常重要的生物学功能。在医药工业中, 以唾液酸为前体的衍生物被越来越多地用于抗病毒药物、治疗神经性疾病药物和诊断试剂等。FDA 和EMA 等监管机构已经对唾液酸在药物开发中的特性提出了要求。此外,唾液酸的含量可以作为批次之间产品一致性的控制手段。因此,开发可靠的唾液酸分析方法变得尤为重要。本文综述了近年来主流的唾液酸分析检测方法,特别关注化学方法和定量分析。
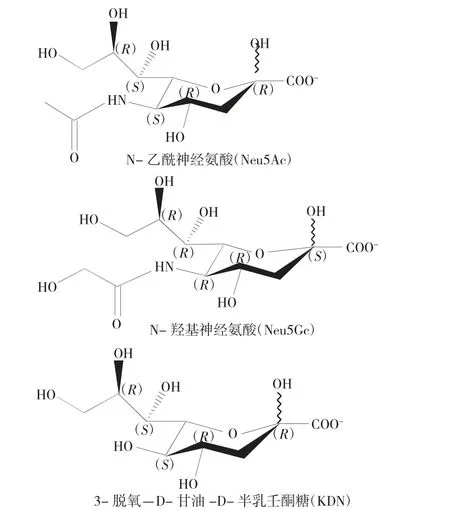
图1 唾液酸的3 种基本形式Fig.1 Three basic forms of sialic acid
1 唾液酸的提取
唾液酸分析的关键步骤是将唾液酸从亲本糖缀合物中分离释放出来。目前,常用的两种释放方法为酸水解和酶水解。在水解程序中关注的焦点是碳水化合物受到最小的破坏和唾液酸的最大释放,许多研究都关注这些条件的优化。
其中,酸水解的应用更为广泛。Ye 等人[3]采用单因素法和响应面法对食品样品的水解条件进行了系统优化,以获得最大含量的唾液酸。提取唾液酸的最佳水解溶液为0.6mol·L-1盐酸。最佳水解温度为80℃,水解时间为14min。
高静[4]对 25mM HCI、25mM 三氟乙酸(TFA)、25mM H2SO4、2M 乙酸等不同酸水解后的糖缀合物中Neu5Ac、Neu5Gc 的含量进行了比较研究。结果表明,HCI 和TFA 的水解效果优于其余酸。
刘志东等人[5]对糖蛋白中唾液酸的水解方法进行了优化和比较。从模型糖蛋白中释放唾液酸效果方面,醋酸水解优于盐酸水解,因为水解副产物生成较少。尽管采用这些酸水解糖蛋白2h , 都将导致20%的损失, 但这能保证唾液酸链的完全水解。
酶水解虽然不能保证所有非游离唾液酸的释放,但酶解法比酸水解法温和,可减少损失。
Nkeonye 等人[6]对生物样品中唾液酸的提取过程包括均质化、酶(磷脂酶和脂肪酶)处理和酸水解。用磷脂酶处理生物样品匀浆导致唾液酸释放增加,最佳酶处理时间为90min,用脂肪酶处理匀浆也导致唾液酸释放量增加,但弱于磷脂酶的作用。而酸水解作用最强,导致唾液酸释放增加15 倍。
2 唾液酸的纯化
由于样品成分的复杂性且唾液酸可以做为蛋白质,脂质和糖类的组成部分,在检测之前往往对唾液酸进行纯化。阴离子交换色谱柱是常用的纯化方法[6],缺点是过程复杂且材料昂贵,适用于复杂样品且其它方法不具备较优的选择性的情况。近年来,分散固相萃取技术使复杂样品中唾液酸的纯化变得简便且易执行。这一技术需选择合适的分散固相萃取的吸附剂对样品进行前处理,目前,利用PBA 及其衍生物与SAs 之间的作用研发吸附剂是热点。
田华军等人[7]在介孔二氧化硅KCC-1 上修饰3-丙烯酰胺基苯硼酸(AAPBA),形成AAPBA@KCC-1材料作为分散相萃取的吸附剂,用于提取和富集血清中的SAs,解吸附后采用傅里叶变换离子回旋共振质谱法(FT-MS)检测血清中的SAs 含量,此方法测定速度快、结果准确。
Huang 等人[8]以 3-氨基苯硼酸(APBA)、甲基丙烯酸缩水甘油酯(GMA)以及Fe3O4为材料制备硼酸亲和磁性空心分子印迹聚合物(B-MhMIP),作为吸附剂在pH 值为4 的条件下吸附唾液酸,在碱性条件下解吸附,再用高效液相色谱法进行测定,在最佳条件下,唾液酸的检出限为0.025μg·mL-1,回收率为70.9%~106.2%,这些结果证实了B-MhMIPs 在选择性高效富集复杂基质中痕量SA 方面表现出优越性,将其推广使用将为SAs 的检测带来便利。
Qu 等人[9]将2-氨基对苯二甲酸连接在锆基金属有机骨架(MOF)上,命名为 UiO-66-NH2,以此作为分散相萃取的吸附剂提取血浆中的唾液酸,然后用5%的醋酸水溶液进行超声解吸附,最后采用高效液相色谱(HPLC)-荧光检测(FLD)相结合的方法进行检测,该方法对生物样品中SAs 的测定具有较高的灵敏度和回收率,检测限低,Neu5Gc 和Neu5Ac的检出限分别为 0.16pmol·L-1和 0.11pmol·L-1,对于分析低浓度的唾液酸有广泛的前景。
Analysis of the possibility of ancient glacier geological relics existing in the middle and low
在最近的一项研究中,提出了一种基于超声作为辅助能量的方法来缩短水解和衍生化的时间和步骤。超声辅助下注射器内封闭水解衍生化(UCSHD)方法除了超声能量的辅助外,还有一个主要的区别特征:水解衍生化是在封闭的注射器系统中进行的。UCSED 技术操作简单、方便、时间较短,与传统管式法相比,有几个额外的优点:(1)无需其他辅助设备,注射器即可准确抽出一定数量的溶液;(2)可以防止挥发和损失,这是准确量化的必要条件,也可以防止对实验人员和环境的潜在威胁;(3)过滤方便。反应结束后,生成的混合物被冷却到室温,在没有额外注射器的帮助下,通过注射器过滤器过滤,这实际上可以避免额外的过滤操作[10]。
3 唾液酸的测定方法
关于唾液酸检测方法的报道有很多,随着科技的发展,早期的硫代巴比妥酸法、薄层色谱法逐渐被更加便捷的高效液相色谱法、传感器法等取代。在HPLC 测定过程中,衍生化试剂与唾液酸反应得到具有荧光特性的化合物,此外,衍生化反应可以避免SAs 解离,提高稳定性,更有利于用色谱法或质谱法进行测定。
3.1 高效液相色谱法(HPLC)-荧光或质谱检测器(FLD/MS)
3.1.1 柱前/后衍生-HPLC 法 王辉俊等人[11]建立了瓜蒌皮注射液中唾液酸的HPLC-MS/MS 定量分析方法。采用多反应监测模式(MRM)对瓜蒌皮注射液中唾液酸进行定量分析。结果说明唾液酸线性关系良好(R≥ 0.995),线性范围为 250~32000nmol·L-1,唾液酸的检出限为3.91nmol·L-1。该方法灵敏性、专属性和选择性好,可用于瓜蒌皮注射液中唾液酸的定量测定和质量控制。
张峰等人[12]建立和验证抗黄斑变性药物康柏西普的3 个质量控制方法,其中包括唾液酸含量的反相高效液相色谱测定方法。间苯二酚法测定唾液酸含量中,N-糖的非唾液酸单糖亦可与间苯二酚反应,产物在580nm 处有光吸收,从而影响检测结果,该研究建立的RP-HPLC 法通过4,5-亚甲二氧基-1,2-邻苯二胺盐酸盐 (4,5-methylenedioxy-1,2-phenylenediamine dihydrochloride,DMB)标记排除该影响,检测结果更准确[12]。
王兰等人[13]初步建立了血管内皮生长因子抑制剂的质控方法和质量标准,通过邻苯二胺(OPD)作为衍生化试剂,采用反向高效液相色谱法进行唾液酸含量分析,为保证该产品安全有效、质量可控打下了一定基础。
伍晓燕等人[14]采用柱前衍生高效液相色谱方法测定燕窝样品与伪品中的唾液酸含量,实验衍生试剂选用的是邻苯二胺盐酸盐溶液,是因为邻苯二胺溶液本身有弱碱性,而唾液酸衍生物在碱性环境下不稳定,因此本实验基于减少显色剂对供试样品的影响,并减少引入其他杂质的考虑,选择邻苯二胺盐酸盐溶液。该方法简单、可靠、准确,可用于鉴别燕窝与银耳、猪皮等伪品,也为判断燕窝质量的优劣提供实验依据。
由于N-乙酰神经氨酸与H2O2可以在体外生成4-乙酰氨基-2,4-双脱氧-D-丙三基-D-半乳糖苷-辛酸(ADOA),为了说明唾液酸类化合物在氧化应激状态下所扮演的角色,KAWASAKI 等人[15]采用DBD-PZ 作为衍生化试剂,建立了亲水作用液相色谱分析-荧光检测方法同时测定唾液和血清中的N-乙酰神经氨酸和ADOA。二者的线性范围分别为221fmol 到 1.5nmol, 44fmol 到 1.5nmol。
NAGAI[16]等人建立了一种测定生物样品中唾液酸的分析方法,并将其应用于胎牛血清、新生小牛血清和成年牛血清中。采用亲水作用色谱分离,柱后与2-氰乙酰胺进行衍生化反应,荧光检测器检测N-乙酰神经氨酸(Neu5Ac)和N -羟乙酰神经氨酸(Neu5Gc)。Neu5Ac 和 Neu5Gc 的校准图在 10pmol-5nmol 范围内呈线性关系。本方法对血清中Neu5Ac和Neu5Gc 的定量有较好的灵敏度。
3.2 表面增强拉曼光谱法(SERS)
表面增强拉曼光谱(SERS)技术是一种基于拉曼光谱发展的可获得物质的分子信息以及具有超高检测灵敏度的光谱技术[17]。为了实现SAs 的特异性检测,通常需要用唾液酸配体PBA 衍生物进行修饰,增强拉曼信号,实现对SAs 的检测。
Teng 等人[18]将金纳米颗粒(AuNPs)、氧化铟锡玻璃(ITO)、4-巯基苯硼酸(4-MPBA)组装在一起,得到ITO/Au/4-MPBA 反应板,能够与SAs 形成可逆的酯化产物。将ITO/Au/4-MPBA 加入到从肺癌患者体内获取的血清中,得到类似与“三明治”类似的夹层结构--ITO/Au/4-MPBA/SA/4-MPBA/Au,最后用拉曼光谱进行检测,可避免糖类物质的干扰,与传统的方法相比,灵敏度和选择性更高。
He 等人[19]用 MPBA 和 4-巯基苯腈(MBN)修饰的银纳米颗粒(AgNPs),制成无背景SERS 纳米探针,可准确定量检测细胞表面的SAs,也可用于监测SAs 在肿瘤细胞表面的动态表达过程,为筛选癌症提供了一种准确的活细胞表面SAs 定量分析方法。
3.3 基质辅助激光解吸/电离飞行时间质谱法(MALDI-TOF-MS)
MALDI-TOF-MS 可用于检测SA,在进行检测前须对样品进行两步衍生化,第一步,先用1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)、1-羟基苯并三唑(HOBt)和二甲胺(DMA)在 60℃的二甲基亚砜(DMSO)溶液中处理糖基化蛋白,以EDC 为羧酸活化剂,HOBt 为催化剂;在第二步中,将等量的氢氧化铵(PH10)加入到后续的反应溶液中,在60℃下保持2h,α2,3-连接的SA 形成稳定的酰胺化结构,而α-2,6-连接的SA 形成稳定的二甲基酰胺,并在氢氧化铵中保持稳定,再采用MALDI-TOF-MS 进行检测,可区分不同键连接的SA。与传统方法相比,此方法可区分α-2,3 与α-2,6 连接的SA,为样品中SA 的检测提供了一种简单而准确的方法[20]。如图2。
3.4 传感器检测法
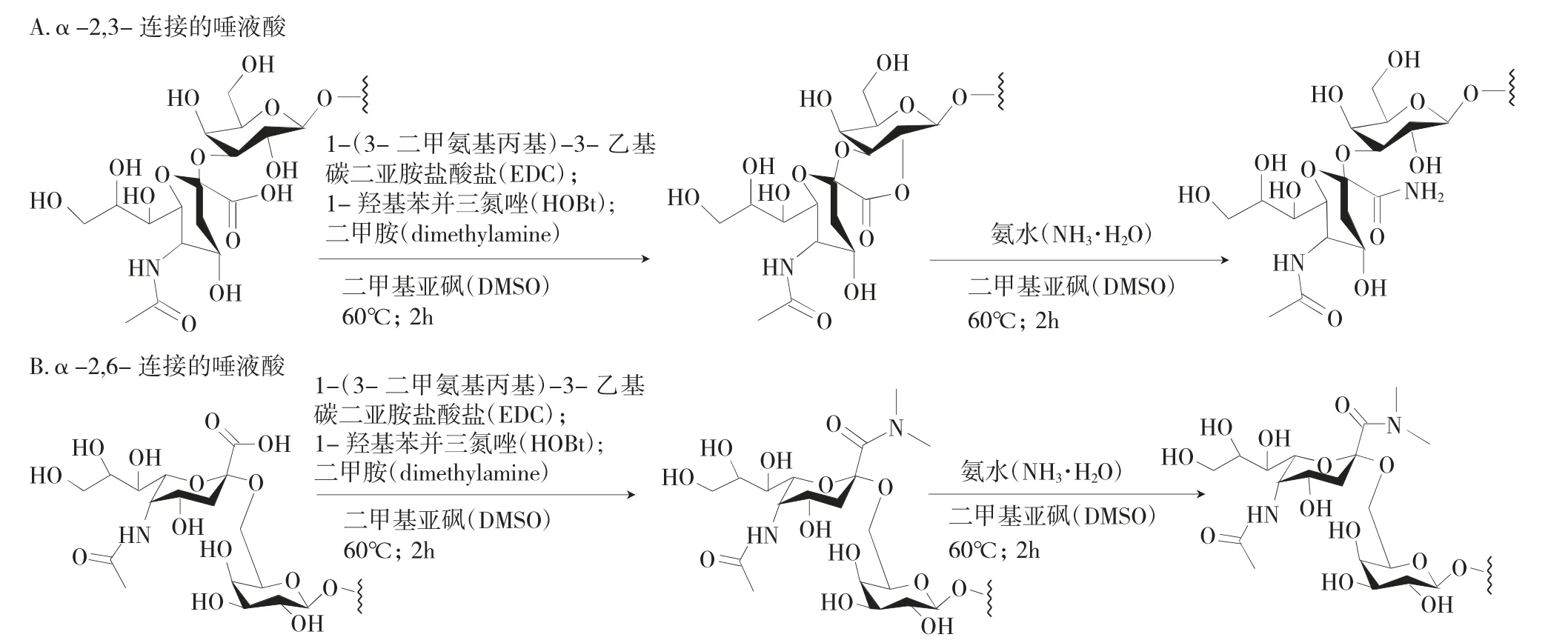
图2 α2,3-连接的SA 以及α2,6-连接的SA 的两步衍生化过程Fig.2 Two-step derivatization process of α2,3-linked SA and α2,6-linked SA
传感器技术是现在的一个研究热点,具有高选择性、高灵敏度和快速的特点。之前研究较多的是酶生物传感器,但是酶生物传感器需要两种酶,物理和化学变化容易影响酶的活性,导致酶难以重复使用,增加了成本,因此,研究者们在寻求更加简单、可靠、高灵敏度的SA 检测和定量方法。
3.4.1 比色传感器 据报道,比色传感器检测法可采用3-氨基苯硼酸(3-APBA)修饰的金纳米颗粒(AuNPs)作为SA 的识别探针。在最佳条件下,在0.15~1.00mM SA 浓度范围内,700 和 520nm 处的吸光度比呈线性增加,检出限为60μM[21]。该方法简单、快速,不需要制备用于糖类比色传感的稳定的硼酸功能化的AuNPs 等繁琐的步骤。
Sankoh 等人[22]基于4-巯基硼酸功能化金纳米粒子(4-MPBA-AuNPs)的聚集,研制了一种简单、选择性的唾液酸检测比色传感器。与唾液酸结合后,溶液由酒红色变为蓝色。该比色传感器具有良好的分析性能,其线性动态范围为80μM 至2.00mM,检测限为(68±2)μM,且不受可能的干扰和样品基质的影响。此外,定量结果仅在10min 内得到。
3.4.2 分子印迹传感器 分子印迹传感器可通过将SA 印迹聚苯胺硼酸(PABA)膜修饰在碳纳米管(CNT)和玻璃碳电极(GCE)上制备而成[23]。该检测策略利用了硼酸-SA 相互作用引起的电化学电位的变化,在生理PH 值下能很好地将SA 与其类似物区分开来,与非分子印迹电极相比,分子印迹电极对SA 有稳定而快速的电化学响应,在SA 的检测中具有潜在的应用前景。此外,研究者们还研制了一种新型分子印迹石英晶体微天平(QCM)传感器,用于选择性测定人体尿样中的SA。研究者们对QCM 芯片的表面进行了修饰,通过烯丙基巯基的自组装在芯片表面引入了可聚合的双键,再将SA 分子印迹聚合物(MIP)纳米膜附着在改性的QCM 芯片表面进行测定。SA 在 0.025~0.50μmol·L-1范围内呈良好的线性关系。该印迹传感器对SA 的检出限为1.0nmol·L-1,同时具有高回收率[24]。在QCM 表面修饰分子印迹聚合物(MIP)是实现样品中SA 高选择性识别的有效途径。
3.4.3 荧光传感器 Wang 等人[25]设计合成了3 种双乙炔(PDA)单体,即 PCDA-Pba,PCDA-Nap 和 PCDA-EA,构建了新型复合PDA 脂质体传感器。采用苯基硼酸(PBA)分子修饰的单体PCDA-pBA 作为SA 识别的受体,采用含有1,8-萘酰亚胺衍生物荧光团的单体PCDA-Nap 作为荧光信号。复合PDA 脂质体形成后,荧光团与共轭主链间的能量转移可抑制荧光团的荧光。存在额外的SA 或SA 丰富的细胞,侧链的构象被扰乱而荧光恢复。方法可用于检测水溶液中的游离SA 以及活细胞表面糖链末端SA的原位检测。
Hao 等人[26]以一种简单而经济的方法合成了一种用于检测SA 的新型苯基硼酸功能化还原氧化石墨烯探针。首先将异硫氰酸荧光素(FITC)和3-氨基苯硼酸(BA)结合得到化合物BA-FITC。其次,添加了还原氧化石墨烯以获取探针。然后用该探针对pH值为7.40 的缓冲液中SA 含量进行分析。该方法可根据正常人血清SA 阈值检测SA 含量是否正常,方法灵敏度高,特异性好,为快速检测SA 提供了一种有吸引力的新思路。
Wang 等人[27]以苯基硼酸(PBA)功能化的Fe3O4/CdTe 磁性纳米粒子为荧光探针,建立了一种检测唾液酸(SA)的荧光新方法。先用氨基修饰Fe3O4纳米粒子,然后将3-巯基丙酸稳定的CdTe 量子点(QDs)与氨基修饰的Fe3O4纳米粒子共价连接,形成Fe3O4/CdTe 磁性荧光纳米粒子。最后将PBA 引入Fe3O4/CdTe 表面,形成PBA 功能化的Fe3O4/CdTe 磁性荧光纳米粒子。这种纳米颗粒能特异性识别SA,荧光强度被SA 淬灭。此外,由于纳米颗粒与SA 的结合物具有磁性,很容易从样品基质中分离出来。在最佳条件下,纳米探针的荧光强度与SA 浓度呈大范围的负线性关系,范围为50~1.50mg·mL-1,检测限为16μg·mL-1。
3.4.4 电化学传感器 Liu 等人[28]基于多巴胺(DA)的指示剂取代法(IDA),首次研制了一种操作简便、高选择性非酶唾液酸电化学传感器。SA 的检测原理是基于指示剂取代法的硼酸-二醇的可逆共价结合,即SA 可以通过置换1,2 -二醇与DA 竞争结合2-氟苯硼酸(FPBA)。该电极由4-羧苯基-氧化石墨烯(TCPP-GO)、DA 和 FPBA 复合材料分别在玻碳电极(GCE)上修饰而成。新型复合材料TCPP-GO 显著提高了电化学传感器的灵敏度。DA 的恢复阳极电流响应与SA 浓度在0.1~7.5mM 的范围内保持良好的线性关系,检测限为28.5μM .该传感器已成功应用于差动脉冲伏安法(DPV)测定人血液和尿液中的SA。回收率为98.0%~104.0%,令人满意。
张珍等人[29]基于血凝素和唾液酸之间的特异性相互作用。首先优化石墨烯的量,然后通过EDC/NHS 活化石墨烯修饰电极表面的羰基,通过共价键偶联将血凝素固定在电极上,构建血凝素电化学生物传感器,通过传感器与唾液酸结合时的变化进行分析检测。该方法对唾液酸的检测范围为10-11~10-17M,检测限为0.837aM,线性相关系数大于0.9,相同浓度(10-9M)的葡萄糖、半胱氨酸、抗坏血酸的干扰很小,4℃保存15d 后,检测RSD 为4.7%,说明该方法抗干扰性强,稳定性好。
Fang 等人[30]基于高效的 Ru(II)复合材料构建了一种硼亲和可再生电化学发光(ECL)生物传感器,用于检测唾液酸(SA)。具体来说,Bi 纳米带(Bi NBs)首次作为共反应物制备了自增强Ru(bpy)2+3-Bi NBs 复合材料,通过缩短电子传输距离和降低能量损失产生强烈的ECL 信号。此外,采用HNO3辅助合成的具备纳米孔性质、单晶结构的金红石TiO2介晶(Nss-TiO2-RM)作为支架,增加了 Ru(bpy)2+3和 Bi NBs 的固定量,进一步增强了ECL 信号。4 -巯基苯硼酸(4-MPBA)作为仿生识别单元可以替代抗体捕捉SA,而且电极可通过0.1M PBS(pH 值为6)完成自我清洁。在最佳条件下,可在 3.5×10-5~350ng·mL-1范围内对 SA 进行灵敏检测,检出限为 1.17×10-5ng·mL-1。
4 结论
过去十多年中,化学衍生化结合现代分离分析方法的快速发展,大大加快了唾液酸及其多糖的研究进展。近年来,兴起的传感器技术大大提高了检测的灵敏度,能够筛查以唾液酸作为潜在生物标记物的疾病,并监测以唾液酸作为靶向的药物的治疗效果,将会有更多的诊断技术和新型药物被开发和应用于医学临床。此外,由于唾液酸在抗炎、抗病毒、抗肿瘤领域的特殊生物活性,科学家们也正着手研发唾液酸相关衍生物,结合唾液酸生物活性的筛选,合成先导化合物,发展快速准确的唾液酸含量测定方法,能够对以唾液酸为有效成分的药物进行更加精确全面的控制,为保证产品质量提供技术保障。
