3,6-二溴-N-烯丙基咔唑化合物的合成探索
2020-09-10郭利安
郭利安

摘 要:3,6-二溴-N-烯丙基咔唑是合成化合物P7C3-A20的中间体,本文主要探索一条3,6-二溴-N-烯丙基咔唑的合成路线,并不断优化反应条件以提高反应产率和选择性。
关键词:P7C3-A20;胺丙基咔唑;溴代
0 引言
P7C3-A20是一种胺丙基咔唑类化合物,AndrewA.Pieper等人[1]于2010年首次发现并报导了此类化合物,它的促神经生成和神经保护功能为神经退行性疾病的治疗提供了可能。本文设计以咔唑为基本原料,经NBS溴化和咔唑N烷基化取代两步反应后得到化合物3,6-二溴-N-烯丙基咔唑。
1 实验部分
1.1 主要实验仪器与试剂
多功能磁力搅拌器(DCG-C型),万分之一天平(BS-224S),旋转蒸发仪(RE-2000A);咔唑(化学纯),N-溴代丁二酰亚胺(NBS)(化学纯),3-溴丙烯(化学纯)。
1.2 化合物合成
1.2.1 3,6-二溴咔唑的合成
在50mL三口烧瓶中依次加入转子、咔唑1g(6mmol,1.0equiv)和分子筛处理过的二氯甲烷20mL,搅拌,待咔唑溶解后加入15g硅胶,充分搅拌15min后取NBS2.13g(12mmol,2.0equiv)以固体分批加入反应中,加入过程中反应体系颜色变为黄色,在室温(25℃)下搅拌30min,其间反应颜色由黄色变为无色,结束反应,用布氏漏斗抽滤除去硅胶,二氯甲烷3×15mL洗涤硅胶,将混合滤液用2×100mL水洗涤,再用50mL饱和食盐水洗涤,用分液漏斗分液后有机层用无水Na2SO4干燥后在减压条件下旋蒸分离出溶剂二氯甲烷,得棕色粉末状晶体。用热的无水乙醇重结晶可得棕色针状纯产品1.05g。母液拌硅胶后以石油醚:乙酸乙酯=30:1作为洗脱剂柱色谱分离,减压条件下旋蒸,干燥后得白色针状产品0.688g,产率89.1%。产物1HNMR(DMSO,400MHz):δ11.62(s,1H),δ8.45
(s,2H),δ7.55(d,1H),δ7.49(d,1H)。
1.2.2 N-烯丙基咔唑的合成
在50mL三口烧瓶中依次加入转子,咔唑1g(6mmol,1.0equiv)和氢化钠0.72g(60%含量,18mmol,3.0equiv)并安装好温度计以控制反应温度。三次抽真空、充氮气保护后加入8mLDMF,在冰醇浴条件下搅拌15min,溶液呈灰褐色浑浊状,其间由鼓泡装置可见有气泡产生。取3-溴丙烯1.45g(2mmol,1.01mL,2.0equiv)于恒压滴液漏斗中,用2mLDMF稀释后缓慢滴加到反应中,控制反应温度在0℃以下。滴加完全后反应30min,停止反应,在冰醇浴下向三口瓶中缓慢滴加水以除去未反应的氢化钠。当停止产生气泡后用二氯甲烷3×15mL萃取反应混合液,再用3×100mL水洗有机层以除去高沸点的DMF,用30mL饱和食盐水洗后用无水Na2SO4干燥,减压条件下旋蒸后得黄色油状液体,常温放置30min后变为黄色玻璃状固体,有特殊刺激性气味,称量得1.188g,产率95.65%。产物1HNMR(CDCl3,400MHz):δ8.13(d,2H),δ7.47(t,2H),δ7.39(d,2H),δ7.25(t,2H),δ6.05-5.96(m,1H),δ5.18-5.03(dd,2H),δ4.92(d,2H)。
1.2.3 3,6-二溴-N-烯丙基咔唑的合成
1.2.3.1 3,6-二溴咔唑与3-溴丙烯的缩合反应
在50mL三口烧瓶中依次加入转子,3,6-二溴咔唑3.25g(10mmol,1.0equiv)和氢化钠1.2g(60%含量,30mmol,
3.0equiv)并安装好温度计以控制反应温度。三次抽真空、充氮气保护后加入20mLDMF,在冰醇浴条件下搅拌15min。取3-溴丙烯2.42g(20mmol,1.7mL,2.0equiv)于恒压滴液漏斗中,用5mLDMF稀释后缓慢滴加到反应中,控制反应温度在0℃以下。滴加完全后反应30min,停止反应,在冰醇浴条件下向三口瓶中缓慢滴加水以除去未反应的氢化钠。当停止产生气泡后向反应液中加入100mL水,搅拌,待反应产物完全析出后过滤,得棕色絮状粗产品,用热的无水乙醇重结晶后干燥可得白色絮状固体3.638g,产率99.67%。
1.2.3.2 N-烯丙基咔唑与NBS的取代反应
在50mL三口烧瓶中依次加入转子、N-烯丙基咔唑1.035g(5mmol,1.0equiv)和分子筛处理过的二氯甲烷10mL,搅拌,N-烯丙基咔唑全部溶解,加入6g硅胶,充分搅拌15min后取NBS1.78g(10mmol,2.0equiv)以固体分批加入反应中,在室温(25℃)下搅拌30min后结束反应,用布氏漏斗抽滤除去硅胶,二氯甲烷3×10mL洗涤硅胶,将混合滤液用2×50mL水洗涤,再用25mL饱和食盐水洗涤,用分液漏斗分液后有机层用无水Na2SO4干燥后在减压条件下旋蒸分离出溶剂二氯甲烷,得棕色粉末状晶体。以石油醚:乙酸乙酯=50:1作为洗脱剂柱色谱分离,减压条件下旋蒸,干燥后得白色絮状产品1.401g,产率76.8%。
产物1HNMR(CDCl3,400MHz):δ8.15(d,2H),
δ7.55(dd,2H),δ8.15(d,2H),δ7.25(d,2H),
δ6.00-5.90(m,1H),δ5.20-4.94(m,2H),δ4.88-4.86(tt,2H)。
2 结果与讨论
从反应收率,产品状态和容易分离提纯的角度出发,合成3,6-二溴N-烯丙基咔唑的较优路线为先对咔唑进行溴代然后再进行咔唑N烷基化反应,下面对3,6-二溴咔唑的合成以及咔唑N烷基化反应的条件进行优化。
2.1 3,6-二溴咔唑的合成
本文通过硅胶法进行合成,该方法具有反应温度低、药品毒性小、产品收率高、催化剂与溶剂可循环使用等优点[2]。
在实验中发现,咔唑在二氯甲烷中的溶解度并不好,改用对咔唑溶解度好的四氢呋喃作溶剂后,溶剂和催化剂硅胶用量明显减少,收率也有所提高(表1)。
2.2 咔唑N烷基化反应
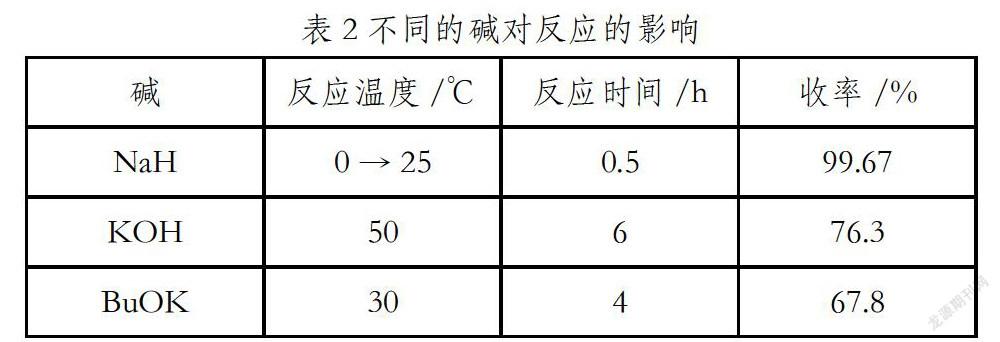
烯丙基溴与3,6-二溴咔唑进行缩合反应,以氢化钠做碱,在溶剂DMF中反应,该反应产率高,反应条件也比较温和,但是钠氢化学性质不太稳定,易变质,加入反应放热也比较剧烈,本文就相同物质的量的不同的碱的反应条件作对比實验,结果如表2所示,当用氢氧化钾或叔丁醇钾作为反应的碱时,两者反应转化率都较低且有副产物生成,尤其是碱性相对较弱的氢氧化钾作为反应的碱时反应需要加热来促进反应进行。所以用钠氢作为反应条件反应效果最好。
3 结论
本文以咔唑为基本原料,先后进行了咔唑的溴代和N-烷基化反应。
采用硅胶法合成3,6-二溴咔唑,以四氢呋喃作溶剂,NBS以固体分批加入反应,常温搅拌30min反应即可进行完全,该反应选择性好,简单柱色谱分离后可得收率为92.3%的纯产品。
3,6-二溴咔唑和烯丙基溴反应合成3,6-二溴-N-烯丙基咔唑,该反应以氢化钠做碱在氮气保护下进行,反应结束后用倒入大量冷水搅拌即可除去反应溶剂DMF而析出产品,用热的无水乙醇重结晶后可得收率为99.67%的纯产品,反应完全且无副产物生成。
参考文献:
[1] Pieper A A,Xie S,Capota E,etal.Discovery of a Proneurogenic,Neuroprotective Chemical[J].Cell,2010(142): 39-51.
[2]李丽荣,崔建兰,蔡留青等.硅胶催化合成3,6-二溴咔唑[J].应用化工,2016,37(6):664-666.
