热休克蛋白5与炎症性肠病研究进展
2020-09-08高菲,范恒
高 菲,范 恒
高菲,范恒,华中科技大学同济医学院附属协和医院中西医结合科 湖北省武汉市 430022
0 引言
炎症性肠病(inflammatory bowel disease,IBD)包括溃疡性结肠炎(ulcerative colitis,UC)和克罗恩病,是一种反复发作的慢性非特异性肠道炎性疾病,其发病机制涉及遗传、环境、肠道屏障和免疫等多个方面,目前并不十分明确.我国现有的流行病学资料统计数据显示,IBD患病率、发病率均呈上升趋势,已成为我国的常见病和多发病.但目前针对IBD的治疗欠佳,大部分治疗方法及药物只能缓解病情,患者需要长期靠药物维持病情稳定,然而疾病仍易复发.因此,寻找新的研究方向以便更有效治疗IBD迫在眉睫.
热休克蛋白5 (heat shock protein 5,HSPA5)又名葡萄糖调节蛋白78 (glucose-regulate d protein78,GRP78)和免疫球蛋白重链结合蛋白,其主要存在于内质网中,属于热休克蛋白70家族,参与多种疾病的发生及发展[1],抑制HSPA5/GRP78对癌症和细菌及病毒感染都有治疗作用,可作为多种疾病的治疗靶点,在IBD治疗上也存在潜在可能[2,3].HSPA5 N端的ER信号序列和C端的KDEL检索序列是区别于其他Hsp70蛋白独特的结构特征,使HSPA5能够转移到内质网并保持内质网蛋白的形式[4].HSPA5具有调控内质网稳态、介导内质网相关细胞凋亡、调控炎症信号通路等与IBD发病相关的作用.有研究表明HSPA5/GRP78 mRNA和蛋白表达水平在IBD患者及鼠的肠组织中显著高表达[5,6],尤其是在具有发达内质网结构的肠上皮细胞(intestinal epithelial cells,IECs)中,当IECs内质网稳态受到破坏时易发生内质网应激[7].但HSPA5在IBD发展过程中具体的作用机制仍不清楚,本文从HSPA5的功能入手,就HSPA5对IBD发病的影响作一概述.
1 HSPA调控内质网稳态
内质网应激(endoplasmic reticulum stress,ERS)主要是指蛋白质合成受到病原体、炎性因子、缺血缺氧等持续性刺激后内质网内大量错误折叠或未折叠蛋白蓄积导致的内质网生理功能紊乱[7,8].HSPA5作为内质网应激的关键因子,能够通过多条途径减少内质网腔内的未/错折叠蛋白,恢复内质网稳态.
未受刺激时,HSPA5与肌醇需要蛋白1 (inositol requiring enzyme 1,IRE1)、蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase,PERK)、活化转录因子6 (activating transcription factor-6,ATF6) 3种应激信号跨膜感受蛋白稳定结合并处于无活性状态;当受到病原体、炎性因子、缺血缺氧等持续性刺激时诱导内质网应激,在内质网中聚集的未折叠或错误折叠蛋白竞争结合内质网分子伴侣HSPA5,使HSPA5与IRE1、PERK、ATF6解离,激活下游信号转导,引发一系列反应来处理错/未折叠蛋白,恢复内质网稳态,这些反应称为未折叠蛋白反应(unfolded protein response,UPR)[9].
首先,当ERS发生时,HSPA5与PERK解离后使之发生磷酸化,其激酶域结构被激活,进一步活化eIF2α,活化的eIF2α在蛋白合成的早期与葡萄糖糖基转移酶及tRNA形成三聚体复合物,从而导致整体翻译停滞,但允许翻译适应性UPR必需的蛋白质[10],从而阻止ERS早期未/错折叠蛋白质的合成.其次,HSPA5一方面可以通过核苷酸结合域和底物结合域两个结构域,直接与未/错折叠蛋白结合使其正确折叠[4];另一方面,HSPA5与IRE1、ATF6解离后使之发生磷酸化,活化的IRE1激活下游因子XBP1形成有活性的转录因子-XBP1s,ATF6活化后释放出N末端片断与通用型核转录因子NF-Y片段结合形成异源二聚体,XBP1s和异源二聚体都作用于ERS元件(ERSE)的特定序列上调HSPA5[11,12],从而正确折叠未/错折叠蛋白.最后,HSPA5参与内质网相关降解(reticulum-associated degradation,ERAD),即错误折叠的蛋白质则被识别后从内质网运输到胞浆中,随后被蛋白酶体泛素化和降解的过程.动物细胞中,IRE1α和IRE1β是IRE1的2种细胞亚型,IRE1α是UPR中的主要参与者,新的研究表明,内质网应激时,HASP5与IRE1α解离,激活IRE1α信号通路,活化XBP1,XBP1s的过度表达增加了ERAD成分Sel1L、Hrd1和Os9的表达[13],促进未/错折叠蛋白的降解;此外,HSPA5负责维持ER在蛋白质转运过程中的通透性屏障,以错误折叠的蛋白质为靶点进行逆行转运,使其能被蛋白酶体降解[14].HSPA5阻止未/错折叠蛋白的合成、使已合成的蛋白正确折叠,促使不能正确折叠的蛋白降解,从而减少内质网内未/错折叠蛋白,恢复内质网稳态,利于细胞存活.
IBD的一个主要特征是杯状细胞病理学,通常被描述为杯状细胞耗竭.杯状细胞是肠粘膜屏障第一道防线黏液层的主要构成,它的耗竭大大减弱了其阻止抗原接触肠腔细胞的能力.MUC2是消化道中的杯状细胞主要分泌黏蛋白,其合成的复杂性使其容易在内质网中错折叠,杯状细胞凋亡由错误折叠的MUC2增加引起活性氧(reactive oxygen species,ROS)水平升高所驱动的,纠正MUC2折叠,抑制活性氧,减轻内质网应激,挽救细胞凋亡[15].有研究发现小鼠的杯状细胞中MUC2蛋白错误折叠,引起HSPA5上调,ERS和UPR被激活,开始时HSPA5代偿性增高以处理错折叠的MUC2蛋白,然而持续的ERS会导致肠黏膜屏障损伤并诱发肠道炎症,其肠道病变表现为杯状细胞缺失、中性粒细胞浸润、肠上皮损伤,与人的UC表现相似,此时HSPA5过表达.杯状细胞对应激源高度敏感,导致它们在早发性结肠炎期间优先凋亡,这可能是使疾病持续发展的早期事件之一.内质网应激早期,HSPA5上调增强MUC2的折叠,有助于缓解杯状细胞的损耗,保持粘膜屏障的完整性,更好地管理IBD[16].
2 HSPA5介导凋亡信号通路
当ERS剧烈持续时,未折叠蛋白反应不能完全代偿缓解细胞损害,为维持内稳态,UPR将激活细胞凋亡信号通路,如C/EBP同源蛋白(C/EBP homologous protein,CHOP)途径、Jun-N-末端激酶(c-Jun-N-terminal kinase,JNK)/p-JNK途径、Caspase途径等[17].
IRE1与GRP78/HSPA5解离后会触发IRE1寡聚化以促进激酶结构域发生自身磷酸化,然后与肿瘤坏死因子受体相关因子2和凋亡信号调节激酶1结合形成复合物,导致JNK下游激活[18].
PERK与GRP78/HSPA5解离后会自体磷酸化和低聚化,elF2α磷酸化能够使ATF4表达增加,使ATF4介导的CHOP活化.ATF6与GRP78/HSPA5解离后被转运至高尔基体,由S1P和S2P两种蛋白酶顺序切割.被切下的ATF6 N端胞质结构域进入细胞核,与ATF/cAMP反应元件SANO和内质网应激反应元件结合,激活靶基因CHOP[19].IRE1途径激活的JNK可以上调CHOP的表达[20].ERS的3条通路都能诱导CHOP激活,而ATF4被认为是主要诱导CHOP转录的因子.
正常状态下Caspase 12与GRP78/HSPA5形成复合体而滞留于内质网表面,使Caspase 12处于无活性状态.当内质网应激持续24 h后,Caspase 12与GRP78/HSPA5发生解离,游离的Caspase 12直接激活Caspase 9继而活化Caspase 3等引起一系列下游级联反应,最终导致细胞凋亡,因此游离的Caspase 12可不经线粒体细胞色素c和凋亡蛋白酶激活因子-1途径直接导致细胞凋亡[21].人类的Caspase4与鼠类的Caspase12是同源蛋白质,二者具有基因序列同源性[22].
细胞凋亡本是ERS过强时的一种保护机制,以清除多余的受损细胞,可减少组织受损,但对于IBD而言,肠上皮细胞凋亡过度形成“凋亡漏”,破环了肠道屏障,使病原体轻易进入肠道引发炎症,未被吞噬清除的凋亡细胞引发免疫调节失常[23-25],伴随数量的减少,IECs抗菌、抗病毒、调节肠道内稳态等功能减弱[7].郭腾飞等[26]发现与空白对照组比较,葡聚糖硫酸钠(dextran sodium sulfate,DSS)诱导的结肠炎组小鼠结肠组织中GRP78/HSPA5、ATF6和CHOP mRNA和蛋白表达水平升高.GRP78/HSPA5主要表达于结肠上皮细胞的胞质及胞膜,ATF6主要表达于结肠上皮细胞的胞质及胞核,CHOP主要表达于结肠上皮细胞的胞核中,表明GRP78/HSPA5-ATF6-CHOP通路相关分子可能在DSS诱导的小鼠结肠炎的发生、发展中起重要作用.沈雁等[27]研究发现与DSS空白组比较,模型组小鼠结肠组织IECs凋亡数明显增多,GRP78/HSPA5、Caspase-12和Caspase-3蛋白表达水平明显升高,GRP78mRNA的表达亦上调.而与模型组比较,小檗碱(Berberine,BBR)组IECs凋亡数明显下降,小鼠结肠组织GRP78/HSPA5、Caspase-12、Caspase-3蛋白表达水平与HSPA5mRNA的表达水平均下调,表明BBR有效减轻UC小鼠的结肠炎症,其机制可能与抑制HSPA5下调ERS水平,抑制ERS介导的Caspase-12/Caspase-3凋亡信号通路活化有关.
3 HSPA5激活核转录因子κB炎症信号通路
应激状态下,HSPA5从内质网上解离和IRE1、PERK和ATF6链接状态,激活这3个基因相关通路及其下游调控的核转录因子κB (nuclear factor-κB,NF-κB)炎症信号通路,引起炎症反应.当应激过度时,HSPA5过表达,炎症反应也相应过度增强,损伤机体组织器官,引起病变.
PERK的激活和伴随的翻译抑制导致核转录因子κB抑制分子(inhibitor of NF-κB,IκB) α合成减少,NF-κB入核增加,短寿命IκB蛋白与长寿命NF-κB蛋白的比例失衡,从而致NF-κB的激活,独立于IκB磷酸化[28].IRE-1与TRAF-2结合后能募集核因子κB抑制蛋白激酶触发IκB激酶和IκB磷酸化.ATF6通过蛋白激酶B磷酸化激活NF-κB.
大量研究已经证实IBD的炎性损伤与NF-κB过度或持续激活密切相关.激活后的NF-κB入核诱导TNF-α、IL-6、IL1β等炎性因子的表达,使得肠道屏障破坏,而这些细胞因子又能够作为NF-κB的刺激剂,可进一步活化NF-κB,造成持续或放大的炎症反应[29].Awada等[30]研究发现长期食用氧化的n-3多不饱和脂肪酸的高脂饲料的小鼠小肠组织HSPA5/GRP78、NF-κB、CHOP的表达显著升高伴随着Paneth细胞数量的减少.可能是由于氧化因素的长期刺激使Paneth细胞处于过度应激状态,GRP78过度表达,激活下游NF-κB、CHOP通路,促使Paneth细胞凋亡.Paneth细胞是肠上皮细胞中重要的分泌细胞,分泌抗菌肽维持肠道菌群动态平衡[31],缺少Paneth细胞,肠黏膜屏障结构破坏,防御病原功能减弱致IBD进展.
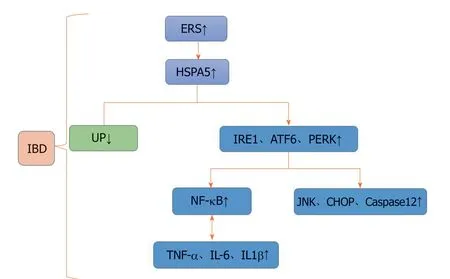
图1 热休克蛋白5在炎症性肠病中影响肠上皮细胞存活及凋亡的机制.ERS:内质网应激;HSPA5:热休克蛋白5;UP:错/未折叠蛋白;IBD:炎症性肠病;IRE:肌醇需要蛋白;ATF:活化转录因子;PERK:蛋白激酶R样内质网激酶;NF:核转录因子;JNK:Jun-N-末端激酶;CHOP:C/EBP同源蛋白;TNF:肿瘤坏死因子;IL:白介素.
4 结论
在内质网应激早期即适应性应激状态下,HSPA5通过3条内质网应激途径代偿性高表达处理内质网的未/错折叠蛋白恢复内质网稳态,从而保护肠上皮细胞,但当应激时间过长或者强度过高即ER过度应激时,HSPA5处理蛋白的负荷过重,它将激活凋亡信号通路和炎症信号通路凋亡通路和炎性通路激活,促细胞凋亡作用占主导.IBD发生时,肠上皮细胞内质网过度应激,HSPA5的致细胞凋亡作用远大于其保护细胞的作用.因此,可以将过表达的HSPA5看作是IBD肠上皮细胞存活的危险因素(图1).目前IBD领域的大多数研究主要集中在产生组织损伤的因子和过程上,而很少考虑细胞保护和细胞修复的内在机制,HSPA5在IBD发病中的分子机制研究尚少,我们需要进一步研究其在IBD肠上皮细胞存活中的相关作用机制,探究其是否可以作为IBD的新的治疗靶点,从而为IBD的治疗提供新的方向.本文仅就内质网中的HSPA5对细胞存活的影响作一阐述,细胞外的HSPA5对细胞存活的影响尚需作进一步研究.
