固相萃取-气相色谱质谱测定牛奶中氯氟氰菊酯和氯氰菊酯残留
2020-09-03吴南村刘春华
张 群,吴南村,马 晨,张 月,刘春华
(1.中国热带农业科学院分析测试中心,海南 海口 571101 ; 2.海南省热带果蔬产品质量安全重点实验室,海南 海口 571101)
一直以来人们极为重视牛奶中兽药、抗生素残留,但我国对牛奶中农药残留的关注不够。我国未制定牛奶中氯氟氰菊酯和氯氰菊酯的最大残留限量[1],欧盟和国际食品法典委员会(CAC)规定了牛奶中氯氟氰菊酯的最大残留限量分别为0.02 mg/kg[2]和0.2 mg/kg[3]; 欧盟和CAC均规定了牛奶中氯氰菊酯的最大残留限量为0.05 mg/kg[4-5]。有报道指出,牛奶中检出氯氟氰菊酯(0.11~0.21 mg/kg)和氯氰菊酯(0.21~0.34 mg/kg)[6],超过欧盟和CAC标准。氯氟氰菊酯和氯氰菊酯作为拟除虫菊酯类农药,这类农药的毒性一般不大,但有蓄积作用,会对人类健康产生长期、微剂量、慢性毒性作用[7-8]。近年来,国内外学者已做了很多这类农药的分析方法探索:气相色谱法[9-12]和气相色谱-质谱法[13-15]、高效液相色谱测定[16-19]和高效液相色谱-质谱法[20-22]等。上述报道主要是标准品或者果蔬、茶、鱼等基质的测定,有关牛奶中这2种农药残留检测的报道较少。本文建立了牛奶中氯氟氰菊酯和氯氰菊酯的固相萃取净化-气相色谱质谱(Solid phase extraction-gas chromatography coupled to mass spectrometry,SPE-GC-MS)检测方法,为监管监测提供技术支撑。
1 材料与方法
1.1 仪器、试剂与材料 7000三重串联四级杆气质联用仪(美国,Agilent公司);EB-280-12电子顶载天平(日本,岛津公司);T25 basic高速匀浆机(广州仪科实验技术有限公司);QL-901旋涡混合器(江苏海门其林医用仪器厂);RE52CS-1旋转蒸发仪(上海亚荣生化仪器厂);C18固相萃取柱(CNW Technologies GmbH,2000 mg/10 mL);毛细管气相色谱柱SLB-5MS(30 m×0.25 mm×0.25 μm,美国SUPELCO公司)。
新鲜牛奶,购于新疆市场。氯氟氰菊酯标准品,纯度≥99%(天津农业部环境质量监督检验测试中心)。乙腈、丙酮、正己烷(HPLC级,美国Fisher);其余试剂均为市售分析纯。
1.2 试验方法
1.2.1 样品前处理 牛奶鲜样500 mL,装入洁净的密封罐中,于-18 ℃编号保存。称取15.00 g牛奶样品,样品中加入20 mL乙腈以及4 g硫酸镁和1 g氯化钠;于振荡器上剧烈振荡10 min,在4 200 r/min条件下离心8 min;收集上清液置于另一个100 mL鸡心瓶中;继续向残渣中加入20 mL乙腈重复提取1次,离心;合并2次提取液,40 ℃水浴旋转蒸发至1 mL左右,待净化。将待净化溶液转移到用10 mL乙腈活化的C18固相萃取柱中,用5 mL乙腈洗涤2次样品瓶,洗涤样品瓶,洗涤液并入固相萃取柱中;收集全部流出液于100 mL鸡心瓶中;用10 mL乙腈洗脱固相萃取柱;合并流出液,40 ℃水浴旋转蒸发至0.5 mL左右;用5 mL正己烷进行溶剂交换,重复操作2次,将收集液旋蒸近干,氮气吹干后,最后定容用正己烷。
1.2.2 气相色谱质谱(GC-MS)检测条件 气相(GC)条件:程序升温,初始温度80 ℃,以35 ℃/min升温至220 ℃,以25 ℃/min升温至250 ℃,以4 ℃/min升温至280 ℃,保持2.3 min;总运行时间:15.0 min;流速:1.0 mL/min;载气:氦气,纯度≥99.999%;进样量:1 μL;进样口温度:260 ℃;进样方式:不分流进样。
SIM条件:电子轰击源:70 eV;离子源温度:230 ℃;GC-MS接口温度:280 ℃,选择离子监测模式(SIM)条件见表1。

表1 SIM模式下待测农药的检测条件Table 1 Detection conditions for tested pesticides in SIM mode
1.2.3 标准溶液及基质标准溶液的配制 标准溶液配制:氯氟氰菊酯和氯氰菊酯用正己烷配制成100 mg/L标准储备液,置于-18 ℃保存。
基质溶液标准品的配制:参照样品前处理方法将空白的样品(未有氯氟氰菊酯和氯氰菊酯检出牛奶样品为空白样品)配制成基质溶液,用基质溶液对标准储备液进行稀释制成所需的定量标准品溶液,现用现配。
1.2.4 准确度与精密度测定 称取4组经测定不含2种农药的空白牛奶样品,每组5个重复。在空白样品中分别添加氯氟氰菊酯和氯氰菊酯的标准品溶液,添加水平为0.01、0.05、0.10 mg/kg和0.20 mg/kg,按照所建立的方法进行检测,计算添加回收率和相对标准偏差(RSD)。
2 结果
2.1 样品前处理条件优化
2.1.1 提取剂的选择 考察了丙酮、乙腈和正己烷作为溶剂提取的效果,结果表明,当添加浓度为0.1 mg/kg时,提取剂为乙腈时,2种农药的样品回收率比较好,故选用乙腈为提取剂(见图1)。
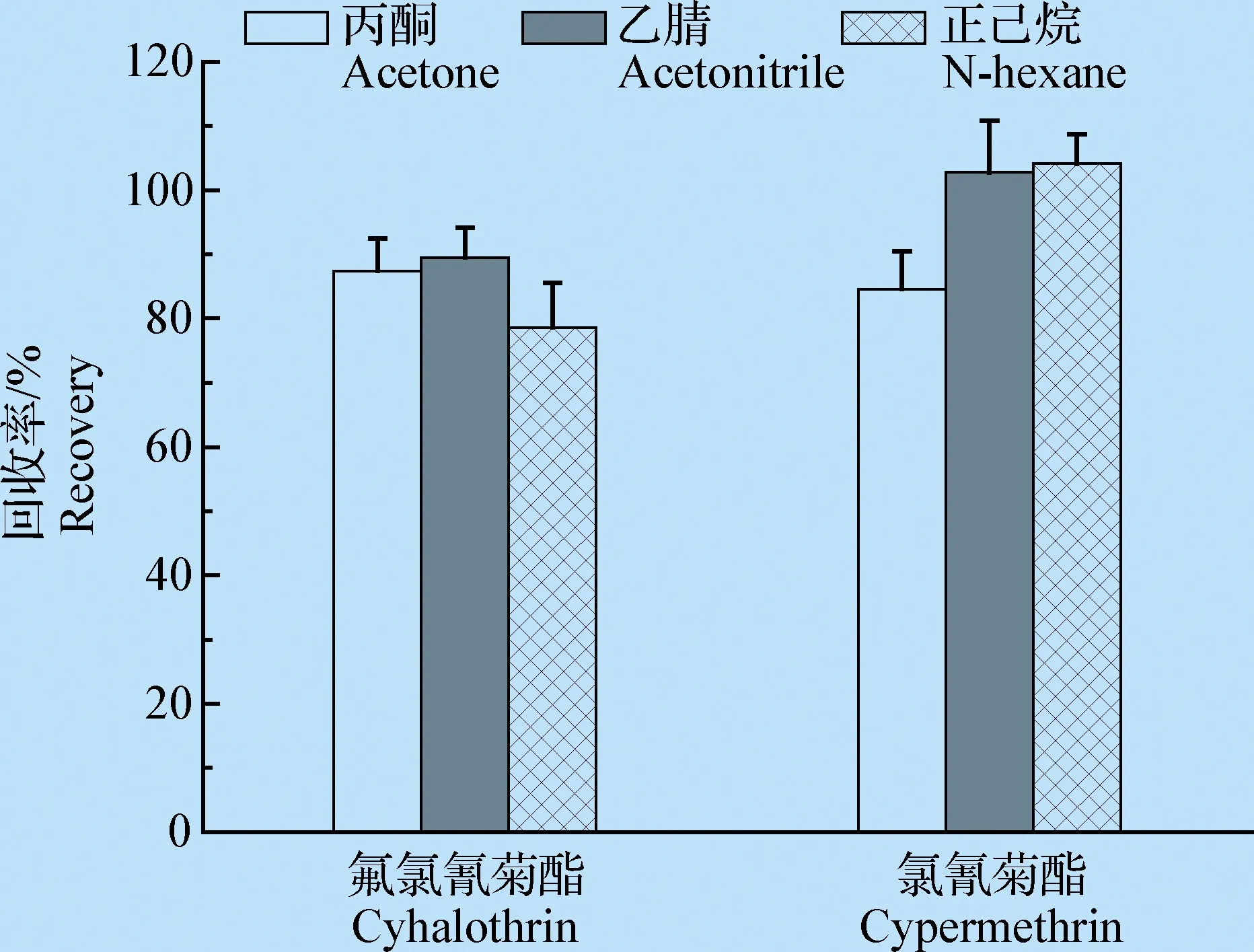
图1 提取剂对2种农药的提取效果(n=5)Fig.1 Extraction effect of extraction agent on 2 pesticides
2.1.2 固相萃取柱的优化 2种农药在3种固相萃取柱[石墨化炭黑(Cabon-GCB)、弗罗里硅土(Florisil)和C18]上均有相当的保留行为,但C18固相萃取柱的回收率较前两者略高,且重复性好。因此本方法选C18固相萃取柱净化。
2.2 待测农药定量和定性离子的选择 首先,在m/z0~450之间分别对2种农药的标准溶液(1.00 mg/L)进行全扫描,确定其保留时间(见表1)和质谱图。在质谱图中,选择特征性、质量数和对称性最高,同时重现性好且不与柱流失碎片离子相同的离子为定量离子。此外,另选择2个特征性、质量数和对称性较高的离子为定性离子。图2为空白样品、基质标准溶液和添加样品在表1条件下得到的多反应监测总离子流图,从图2中可以看出,2种农药所得到的谱峰峰形尖锐,对称性好,各谱峰间完全分离。
2.3 线性方程、定量限、回收率和精密度
2.3.1 线性方程 按1.2.3所述配制的0.01~1.00 mg/kg内5个梯度浓度的混合基质标准品进样,浓度分别为:0.01、0.05、0.1、0.2 mg/L和1 mg/L。在1.2.2的检测条件下,以进样浓度(mg/L)为横坐标,峰面积为纵坐标,绘制线性方程(表2)。结果表明,在0.01~1 mg/L范围内峰面积与进样浓度呈良好的线性关系,决定系数R2>0.99。
2.3.2 定量限 依照3倍信噪比(S/N)对牛奶中2种农药的检出限(LOD)进行计算,确定为9×10-5mg/L;再依据10倍信噪比确定其定量限(LOQ)为3×10-4mg/L。通过实际添加回收试验,最终确定牛奶中氯氟氰菊酯的定量限为0.005 mg/L,氯氰菊酯的定量限为0.01 mg/L,此时5次试验的平均回收率分别为87.3%和92.0%,相对标准偏差(RSD)分别为8.2%和3.5%。
2.3.3 回收率和精密度 根据牛奶中氯氟氰菊酯和氯氰菊酯的最大残留限量值以及方法的灵敏度,对牛奶样品设定了0.01、0.05、0.10 mg/kg和0.2 mg/kg四个浓度的添加水平,牛奶中的添加回收率与相对标准偏差见表3。在0.01、0.05、0.10 mg/kg和0.20 mg/kg四个添加水平下,氯氟氰菊酯和氯氰菊酯残留的平均回收率在83.1%~102.8%;5次重复试验测定的相对标准偏差范围为1.4%~9.1%,均满足定量分析要求。
2.4 实际样品的检测 应用此方法对市场销售的实际样品进行检测,选购的15份样品中均未检出这2种农药残留。
3 讨论
牛奶中脂肪、蛋白质含量较高,基质复杂[23],要对牛奶中的农残进行提取,然后净化样品有效的提取目标物。现有文献及标准中常用丙酮、乙腈和正己烷等作为提取剂提取动植物中的农药残留[1,9-10,19-20],本试验考察了丙酮、乙腈和正己烷作为溶剂提取的效果,发现提取剂为乙腈时2种农药的添加回收率比较好,选择乙腈为提取剂。这一结果可能是乙腈与水部分互溶,加入氯化钠后,盐析、分层和除杂效果明显。然后,考察了拟除虫菊酯类农药残留检测常用的3种固相萃取柱[24-28]-石墨化炭黑(Carbon-GCB)、弗罗里硅土(Florisil)和C18的净化效果,2种农药在3种固相萃取柱上均有相当的保留行为,但C18固相萃取柱的回收率较前两者略高,且重现性好,本方法选择C18固相萃取柱净化。这可能归功于以硅胶为基质的反相C18萃取柱强疏水性选择性,适用于从水性样本中保留大多数有机化合物,同时其具有高键合密度,低流失,高回收率等特点,可以更有效地去除牛奶中脂肪和脂类等干扰物影响而保留目标物得到更高的回收率,且重现性好。
GC-MS的全扫描模式,由于扫描范围宽、频率低,导致背景值基线较高,会影响目标农药的灵敏度。本方法采用选择离子监测模式,可以有针对性地选择目标化合物的特征离子监测,提高了方法的灵敏度,降低了干扰,每种化合物选择3个特征离子,根据目标农药的保留时间和对应特征离子的丰度比,可对样品中存在的目标农药进行定性,可排除在复杂基质中与农药化合物保留时间一致而不是农药的化合物的假阳性结果。如果测定样品检出的色谱峰保留时间与标准样品一致,并在扣除背景后的样品质谱图中,所选择的离子均出现,而且所选择的离子丰度比与标准样品的离子丰度比相一致(相对丰度> 50%,允许± 10%偏差;相对丰度>20%~ 50%,允许±15%偏差;相对丰度> 10%~20%,允许±20%偏差;相对丰度≤10%,允许±50%偏差),则可判断样品中存在这种农药[29]。García-Rodríguez等利用固相微萃取(SPME)方法结合GC-MS测定水产养殖海水样本中的有机磷和拟除虫菊酯农药,该方法具有良好的线性度、精度和较低的LODs[15]。杨丽莉等采用C18固相萃取柱富集环境水中六六六、滴滴涕和环氧七氯,用少量有机溶剂洗脱,添加内标后利用GC-MS的SIM模式进行目标农药的定性、定量,干扰小,测定灵敏度高[30]。本方法定量选择一个干扰小、特征性高的离子,采用标准曲线外标法通过工作站进行,结果同样表明,线性响应和精密度良好,准确度和灵敏度高。

表2 2种农药的线性方程及决定系数Table 2 Linear equations,determination coefficient and linear ranges of the two pesticides
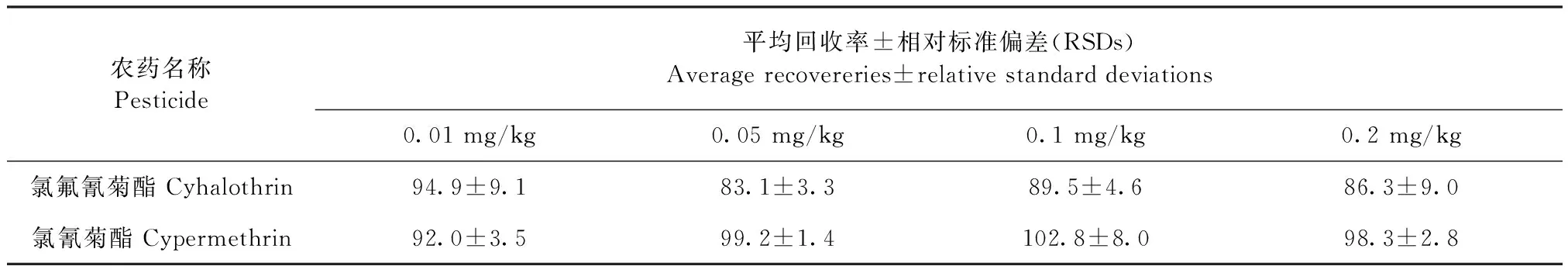
表3 2种农药的加标回收率及其相对标准偏差Table 3 Average recovereries and relative standard deviations (RSDs) of the two pesticides (%,n=5)
长期以来国内外学者对牛奶中兽药残留研究较多,特别是抗生素,牛奶中农药残留的研究较少。国内外毒理学研究专家研究表明,牛奶中农药残留污染对人体健康存在微剂量、慢性毒性[7-8,31]。本试验建立了C18固相萃取柱净化的气相色谱质谱法测定牛奶中氯氟氰菊酯和氯氰菊酯残留的检测方法,样品前处理简便快速且回收率高,能够较好地排除基质干扰;定量限及精密度均能满足痕量分析的要求,而且可以满足欧盟等国牛奶中氯氟氰菊酯和氯氰菊酯的最大残留限量的检测要求,为其在畜禽养殖中违禁使用的监管监测提供理论依据和技术支撑,保障人类健康,回避潜在食用风险。
