以孤立肌肉病为首发症状的Danon病2例临床分析
2020-09-01张岩常杏芝姚玲
张岩, 常杏芝, 姚玲△
1天津市第五中心医院儿科(天津 300450); 2北京大学第一医院儿科(北京 100034)
Danon病是一种罕见的X连锁显性遗传性疾病,由Danon在1981年首次报道,因本病特征性病理改变为骨骼肌和心肌细胞内存在自噬空泡和糖原颗粒增多,故曾被称为X连锁的空泡性骨骼肌心肌病或酸性麦芽糖酶正常的溶酶体糖原贮积症[1]。该病的致病基因为编码溶酶体相关膜蛋白2(lysosomal-associated membrane protein 2, LAMP2)的LAMP2基因。该病特征性临床表现为心肌肥厚、骨骼肌病变和智力发育迟缓[2-4]。Danon病的首发临床症状多为心脏症状,以孤立肌肉病为首发表现者临床罕见。现报道2例基因确诊的以孤立性肌肉病为首发症状的Danon病,以丰富LAMP2基因突变的临床表型,提高对该病的认识,以改善患者的预后。
1 资料与方法
1.1 病例资料收集 收集2016年1月至2017年6月本院基因确诊的2例Danon病患者的临床资料,包括详尽的病史询问和系统的体格检查、常规血液生化检测、血氨基酸及肉碱谱分析、尿有机酸筛查、心电图、超声心动图、肌肉活检病理及基因检测资料。
1.2 临床资料 例1,男,9岁5个月,主因“发现肌无力4年余”入院。隐匿起病,家长无意中发现患儿行走不稳,易摔跤。院外查头颅核磁共振未见异常,心电图正常。患儿为G1P1足月顺产儿,起病前智力运动发育同正常同龄儿童。例2,男,4岁10个月,主因“肢体乏力10月余”就诊。患儿为G1P1足月顺产儿,起病前智力运动发育里程碑基本同正常同龄儿童。既往史及家族史无特殊。
1.3 基因分析 两例患者及其家系成员均采取外周血DNA,应用基因靶向捕获二代测序技术进行了遗传性肌病基因包(gene panel)筛查,参考基因组版本为GRCh37/hg19。对可疑致病基因变异应用Sanger一代测序技术进行验证并进行保守性分析,并通过PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、MutationTaster(http://www.mutationtaster.org/)、GERP++(http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html)进行基因变异的致病性预测(北京康旭医学检验所)。
1.4 随访 电话或门诊随访患者2年,包括肌无力情况、智力运动发育和心脏情况等。
2 结果
2.1 入院体格检查 例1:双眼未见水平眼球震颤,心肺腹(-),双上肢肌力正常,双下肢肌力Ⅴ-、肌张力正常,肌容积小。四肢深腱反射均对称引出,余(-)。例2:神志清,可正常交流,体型消瘦,心肺腹(-)。颅神经(-),四肢肌力、肌张力正常,肌容积少,四肢深腱反射均对称引出,余(-)。
2.2 辅助检查 两例患者血、尿、便常规,血氨基酸和肉碱谱分析及尿有机酸筛查均未见异常。血乳酸、血氨和同型半胱氨酸正常,例1血肌酸激酶400~1 400 IU/L,例2肌酸激酶356~780 IU/L(正常值0.5~25 IU/L)。心电图和超声心动图均未见异常。例1左肱二头肌活检见肌纤维内嗜碱性颗粒物质和不规则空泡(图1)。超微病理检查示肌纤维内可见自噬性空泡和肌纤维膜下糖原颗粒物质堆积,未见结构异常线粒体和类结晶样包涵体(图2)。
2.3 基因突变分析 两例患儿均证实存在LAMP2基因c.877C>T(p.Arg293Ter)变异,该变异导致编码第293号氨基酸Arg的密码子变为终止密码子,从而使肽链合成提前终止,为文献已报道的致病无义变异[5]。例1为新生变异。例2父亲该位点未见异常,其无症状母亲为杂合子。见图3~6。
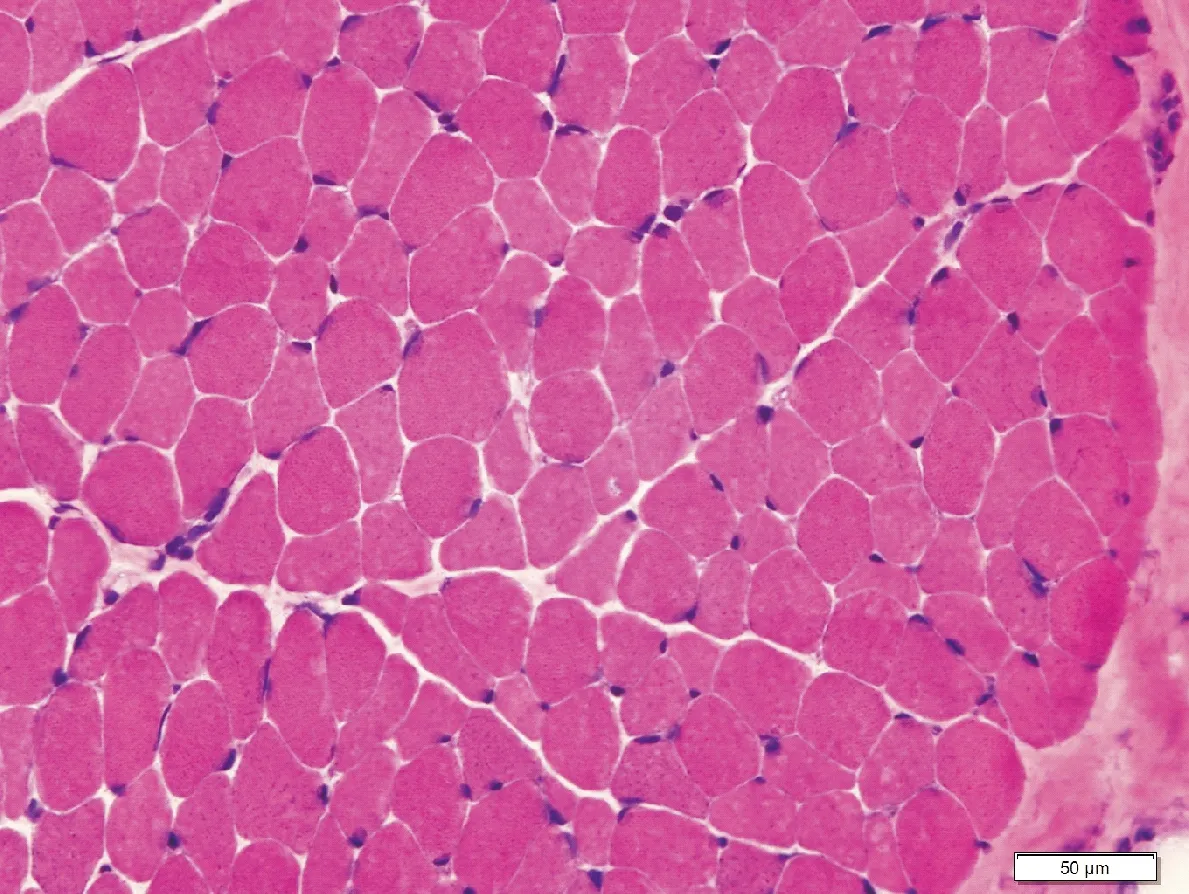
图1 肌纤维内可见一些不规则小空泡和嗜碱性颗粒(HE,×200)
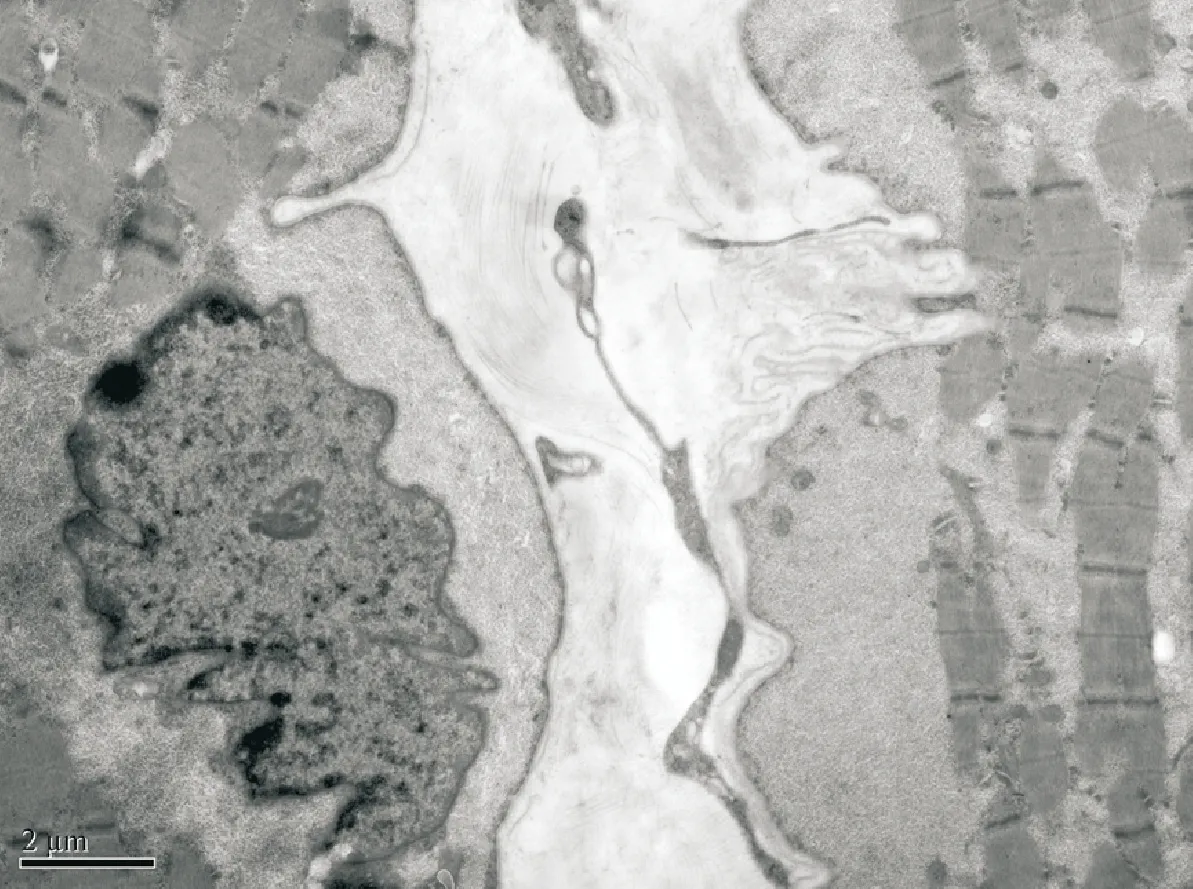
注:胞浆内存在许多膜性自噬小体,其内含有糖原颗粒和胞质降解后的碎片
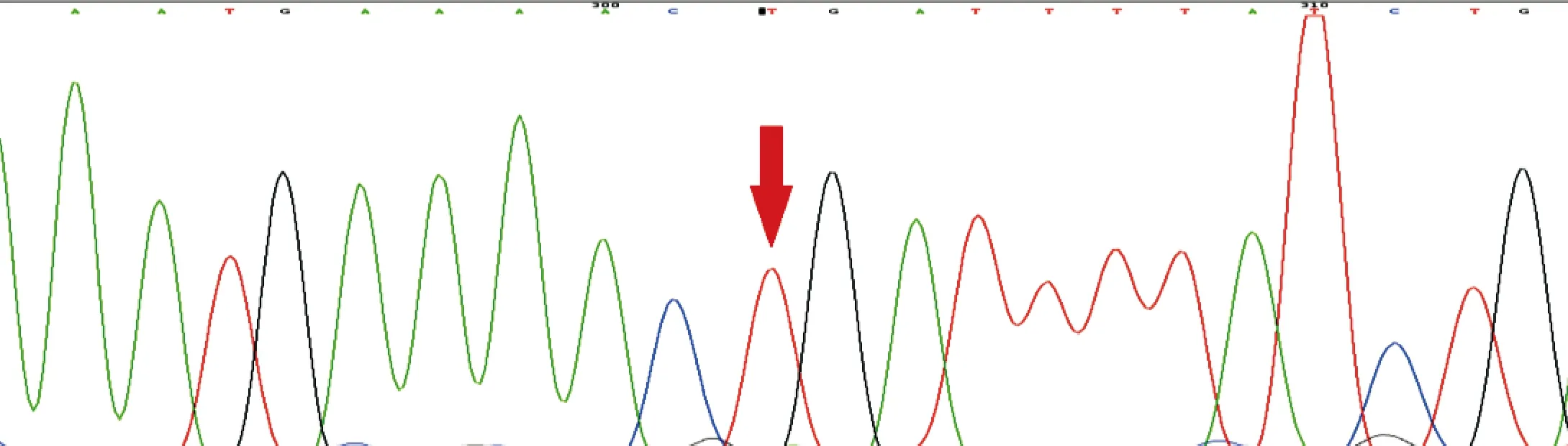
注:箭头示LAMP2基因c.877C>T(p.Arg293Ter)
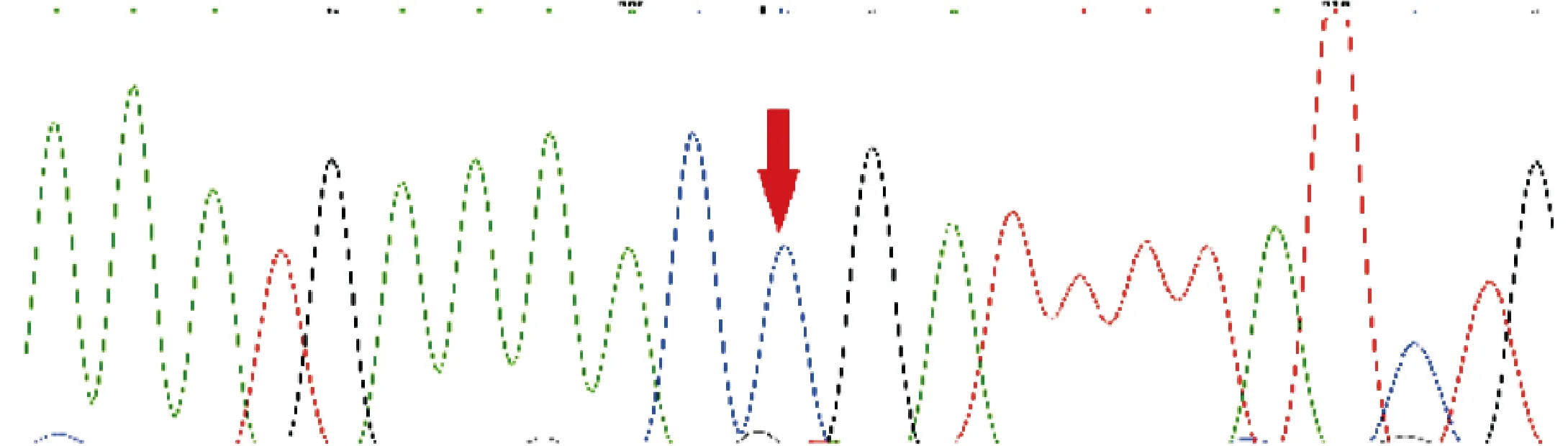
图4 例1父亲的LAMP2基因测序图
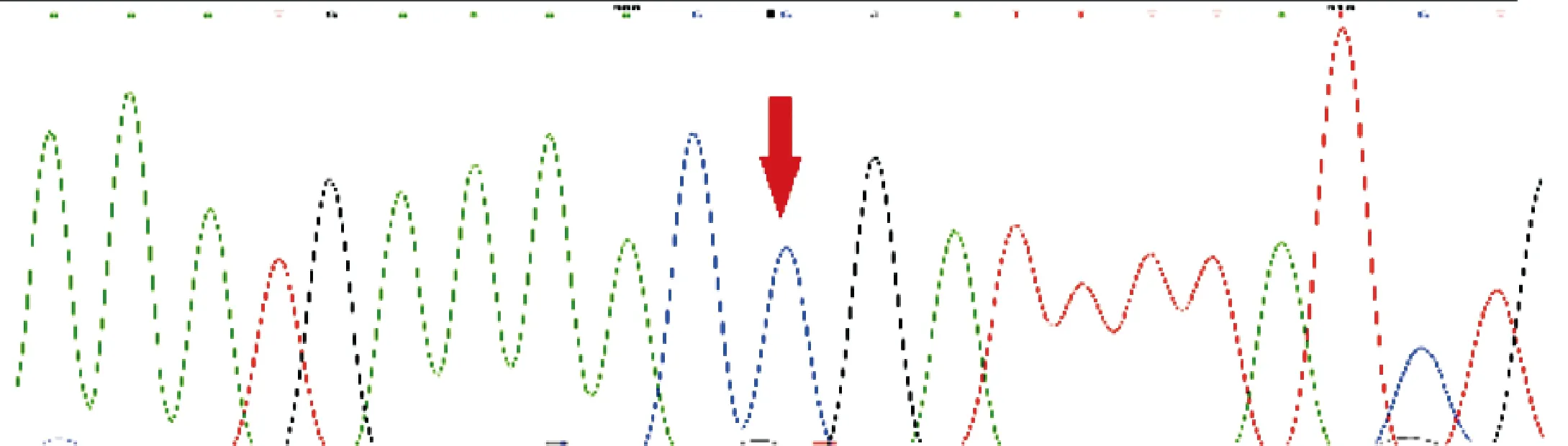
图5 例1母亲的LAMP2基因测序图
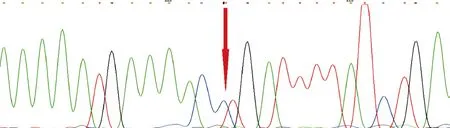
图6 例2母亲的LAMP2基因测序图
2.4 随访 确诊后10个月随访,两患儿无心肌病症状,复查心脏超声心动图和心电图均未见异常。智力发育评估基本同正常同龄儿童。例2母亲30余岁仍无临床症状,建议行超声心动图检查被拒绝。两年后随访,例1患儿运动不受影响,仅运动后多汗,动态心电图示PR间期<0.12 s,胸前导联可见δ波。复查超声心动图仍未见异常。例2失访。
3 讨论
Danon病是一种罕见的X连锁显性遗传性疾病,由位于Xq24上的LAMP2基因突变所致。男女均可发病,但男性较女性患者发病早、进展快、预后差。男性的发病年龄为10个月至19岁,常在30岁之前死亡,平均死亡年龄约(19±6)岁;而女性发病年龄为12~53岁,平均死亡年龄为(40±7)岁[6]。本病特征性临床表现为心肌肥厚(88%)、骨骼肌疾病(80%)和智力发育迟缓(100%)。Danon病女性杂合子(即携带者)也可能出现临床症状,其中6%~47%的患者可能发生认知障碍,61%~100%有心肌病的证据,33%~50%可以有骨骼肌无力[7]。在所有患者中,心肌病变常常是首发症状,表现为较大活动量时胸痛、胸闷、气促,有时伴头晕、黑矇,休息后可缓解。症状逐渐加重,运动耐力下降,有时出现活动时晕厥[8]。心肌肥厚,尤其是左室心肌肥厚是影响患者预后的最主要因素,患者死亡的主要原因为严重心力衰竭和心律失常[6]。文献报道心律失常可见于86%~100%的患者,其中WPW(Wolf-Parkinson-White)综合征是最常见的心电图表现,约占69%。因此,有学者认为在肥厚型心肌病的年轻男性中,WPW综合征的存在强烈提示Danon病[9]。Danon病的其他症状,包括肌肉病、智力残疾和眼部疾病通常被认为是轻微的,不危及生命[9]。多数患者无肌肉病症状,多因心肌病就诊时检查发现肌酸激酶显著升高,进一步检查才发现肌肉受累。本组2例患儿与既往报道不同,均以孤立的肌肉病症状起病,随访2年,目前尚无心脏病受累症状,仅例1患儿心电图有轻度PR间期缩短。
Danon病的诊断主要依据特征性临床和病理表现,结合LAMP2基因检测以明确。因为LAMP2基因编码的溶酶体相关膜蛋白2(LAMP2)是溶酶体膜的组成成分,其三种不同的剪接异构体LAMP 2A、LAMP 2B和LAMP 2C均在细胞自噬通路中发挥重要作用[10-11],因此,Danon病特征性病理改变是心肌和骨骼肌细胞内可见自噬空泡[12-14]。虽然这些自噬空泡内的物质PAS染色阳性,但酸性麦芽糖酶活性正常,这也是Danon病既往被称为酸性麦芽糖酶正常的溶酶体糖原贮积症的原因。基于DNA检测的非侵入性和二代测序技术的应用,LAMP2基因检测是目前确诊Danon病最常用的方法。骨骼肌细胞免疫组化染色显示LAMP2蛋白缺乏,有助于辅助判断LAMP2基因变异的致病性。本组例1患者骨骼肌活检可见骨骼肌内自噬空泡和糖原颗粒堆积,符合Danon病典型病理改变。两例患者均发现已报道的LAMP2基因c.877C>T(p.Arg293Ter)致病变异,为LAMP2基因变异所致的Danon病诊断明确。
Danon病的临床表型与基因型间的关系尚不明确。自2000年Nishino等[3]首先明确LAMP2基因突变为Danon病的致病原因后,多种不同的基因突变相继被报道。目前为止,有大约75个LAMP2突变被报道,其中c.928 G> A被认为是最常见的突变。常见的致病突变类型是无义或移码突变,也有剪接、大片段缺失或重复、插入/缺失和错义突变[7]。关于基因型与临床表型的关系:(1)不同基因突变类型可能影响临床表型。D′souza等[9]认为无义、移码和大片段缺失/重复变异患者的发病年龄较早,而剪接突变的患者发病年龄较晚。本组2例患儿均为LAMP2基因c.877C>T(p.Arg293Ter)为无义突变,发病年龄分别为4岁和5岁,均较早,符合D′souza等的观察。(2)相同的基因突变位点在不同人种间临床表现可能不同。既往文献报道的LAMP2基因c.877C>T(p.Arg293Ter)位点突变的西班牙裔患者是以心肌病症状起病的,12岁时诊断为肥厚型心肌病,不伴明显肢体无力[5],而本组2例患儿均以孤立的肌肉病症状起病。(3)在相同人种甚至同一家系内,相同基因位点在不同性别之间的临床表现也存在差异。文献报道一个西班牙家系,先证者为7岁男孩,病前发育正常,7岁开始出现精神运动发育迟滞,运动不耐受,血CK升高。而患者母亲儿童期无症状,无智力发育迟缓,成年后患产后扩张型心肌病,并接受了心脏移植手术。分子遗传学检测证实患儿及其母亲均存在LAMP2致病变异[15]。
Danon病目前尚无特效治疗方案,预后不良。Danon病与其他肥厚性心肌病不同,患者的心肌病变往往进行性加重,迅速发展到终末期,往往需要心脏移植。患者在童年时常处于无症状期,随后在青春期进入疾病快速进展期,最后死于暴发性心脏疾病[16]。现有数据表明,大多数Danon病患者的自然寿命不超过30年[16]。因此,早期、及时诊断和进行心脏移植的评估对于改善预后至关重要[17]。本组2例患儿目前均无心脏受损征象,但仍需密切随访。尤其是例1患儿5岁发病,4年后确诊为Danon病,随访两年时发现动态心电图PR间期缩短和胸前导联见δ波。目前患儿已11岁,我们将缩短随访时间,密切观察心脏情况。本组患者例2的母亲为杂合子,虽然30余岁尚无临床症状,仍需密切监测。遗憾的是患儿母亲在就诊后10个月随访时拒绝接受心脏彩超和心电图检测,之后就失访了。
临床应重视以孤立肌肉病为首发表现的Danon病患儿,尽早行分子遗传学检测以明确诊断。对于确诊患儿,应严密监测患儿的心脏功能,以改善预后。对患儿家族中女性成员应进行基因变异筛查,对无症状杂合子女性应长期随访并提供生育咨询。
