大豆谷丙转氨酶的结构模型构建及其应用
2020-08-19钟镕华孟繁梅郑晓如伍俭儿张尚宏
林 深, 钟镕华, 孟繁梅, 郑晓如, 伍俭儿, 张尚宏
(1. 中山大学 生命科学学院, 广州 510275; 2. 中山大学 生物工程研究中心, 广州 510275)
谷氨酸-丙酮酸氨基转移酶(Glutamate-pyruvate transaminase, GPT),简称谷丙转氨酶,又称丙氨酸氨基转移酶(Alanine aminotransferase),是自然界中广泛存在的一种酶,在维持生物正常的葡萄糖和氨基酸代谢中起到重要的作用,更是人体肝病、糖尿病和冠心病等疾病的重要预测因子[1]。研究表明,谷丙转氨酶皆属于吡哆醛-5-磷酸(Pyridoxal-5-phosphate, PLP)依赖性酶家族,而吡哆醛-5-磷酸是一种多功能的辅助因子,辅助酶催化氨基酸的各种反应,包括转氨、脱羧、外消旋、β-和γ-消除和取代、羟醛缩合反应和Claisen反应[2-3]。有研究表明,在细菌的氨基酸代谢中最重要的部分是谷丙转氨酶所催化的代谢反应,通过丙氨酸和谷氨酸的相互转化,精确控制丙氨酸和谷氨酸浓度[4]。
尽管GPT的催化机理已经得到了阐释,但目前对于植物来源GPT的研究很少,三维结构明确的植物GPT更是只有大麦(Hordeumvulgare)来源的GPT(PDB登记号3TCM)[5]。分子模型的构建是深入研究蛋白质各项性质的基础,构建一个新的植物GPT的模型,将对其他同类型酶的研究提供便利与支持。由于解析晶体难度大、耗费高,利用相似度高的晶体结构进行同源建模成为一种被广为接受且有较高准确性的方法[6]。使用软件模拟的方法,得到蛋白质三维结构模型,探究模型在不同浓度的缓冲溶液中的构造变化,可以初步得到蛋白质的一些性质。
另一方面,近年来,亲和标记被广泛应用于蛋白质纯化,例如聚精氨酸标签(Poly-Arginine tag, Arg-tag)、钙调蛋白结合肽(Calmodulin-binding peptide)、纤维素结合域(Cellulose-binding domain)、蛋白质二硫化物异构酶I(Protein disulfide isomerase I, DsbA)、聚组氨酸标签(Poly-Histidine tag, His-tag)、Flag标签(Flag-tag)、链霉亲和素结合肽标签(Streptavidin-binding peptide, SBP-tag)等[7]。其中组氨酸标签的应用最为广泛,PDB中接近25%的蛋白质是组氨酸标记后,通过金属固定化亲和色谱纯化得到的[8-9]。然而,组氨酸标签在表达时可能被包埋于蛋白质中而无法被吸附剂吸附,且组氨酸标签可能会影响酶的构象[10],因而在亲和层析前预测组氨酸标记后蛋白质的结构,能够预判用组氨酸标记纯化蛋白的可行性。
大豆(Glycinemax)作为一种常见的固氮农作物,其材料易得,经济效益高。在对大豆的研究中发现GPT存在于大豆的所有组织中[11],然而目前还没有关于大豆谷丙转氨酶(GlycinemaxGlutamate-Pyruvate Transaminase,GmGPT)三维模型的研究。本文从软件模拟建模的角度,探究GmGPT的结构特征和活性区域及其拓展应用,为大豆乃至植物来源GPT的研究提供更多的理论准备。
1 材料与方法
1.1 大豆谷丙转氨酶序列的获得
在NCBI数据库里查询已知的GmGPT的可能序列,共得到了7个已知登录在数据库中的序列,如表1。
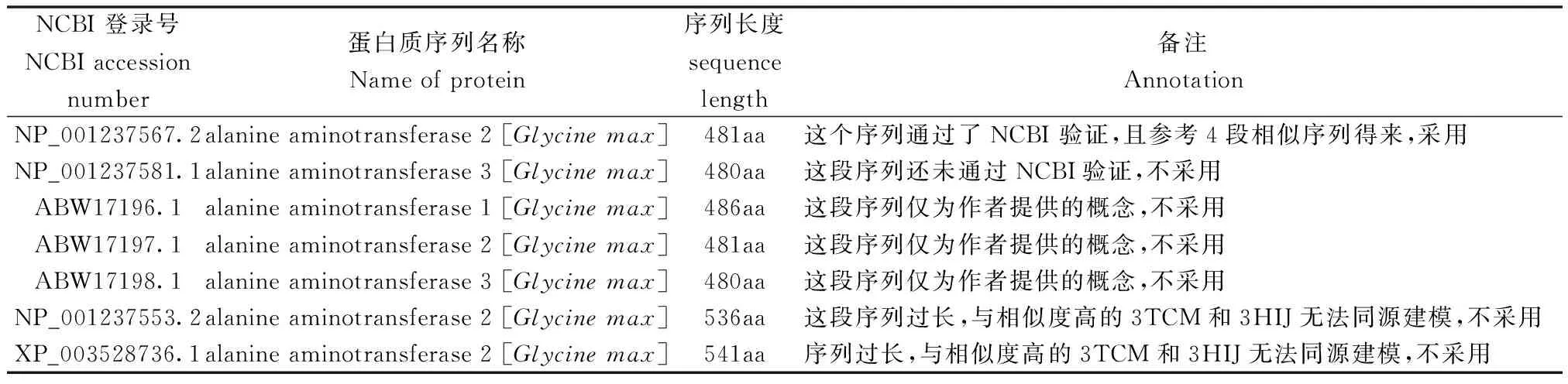
表1 NCBI的protein数据库中大豆的谷丙转氨酶的序列
考虑到序列准确性和建模的正确性,我们挑选了经过验证且参考序列较多的NP_001237567.2序列作为我们建模的出发序列,其序列如图1。

图1 NP_001237567.2的序列
1.2 Blast搜索,多重序列比对,同源建模
在NCBI的blast程序搜索与NP_001237567.2序列相似度高且已知晶体结构的蛋白质,得到对应的大麦GPT(PDB登录号3TCM)和人源GPT(PDB登录号3IHJ),序列相似度均大于40%,符合同源建模要求。利用Discovery Studio的Align Multiple Sequences模块,以BLOSUM矩阵作为多重序列评分矩阵,综合比对出发序列和3TCM序列以及3IHJ序列。对比结果如图2。利用Discovery Studio(版本号2.5)的Build Homology Models模块,以3TCM和3IHJ为模板构建大豆谷丙转氨酶模型。经过简单的loop修饰得到模型A,并通过UCLA的Saves平台对模型A的各项建模指标进行验证。
1.3 模型A在溶液中的状态模拟
应用Discovery Studio(版本号2.5)的Solution模块将模型A分别置于浓度为0.05、0.10、0.15、0.20、0.25和0.30 mol/L的KH2PO4-Na2HPO4模拟缓冲溶液中(pH 7),依次采用Steepest Descent及Conjugate Gradient算法,分步采用固定蛋白质整体、蛋白质主链及全部柔性处理,对模型能量最小化[12]。通过UCLA的Saves平台对各个模型的各项建模指标进行验证,取其中指标最好的一个模型作为GmGPT的溶液状态模型B,并对此模型进行活性位点与酶反应底物对接的分析。
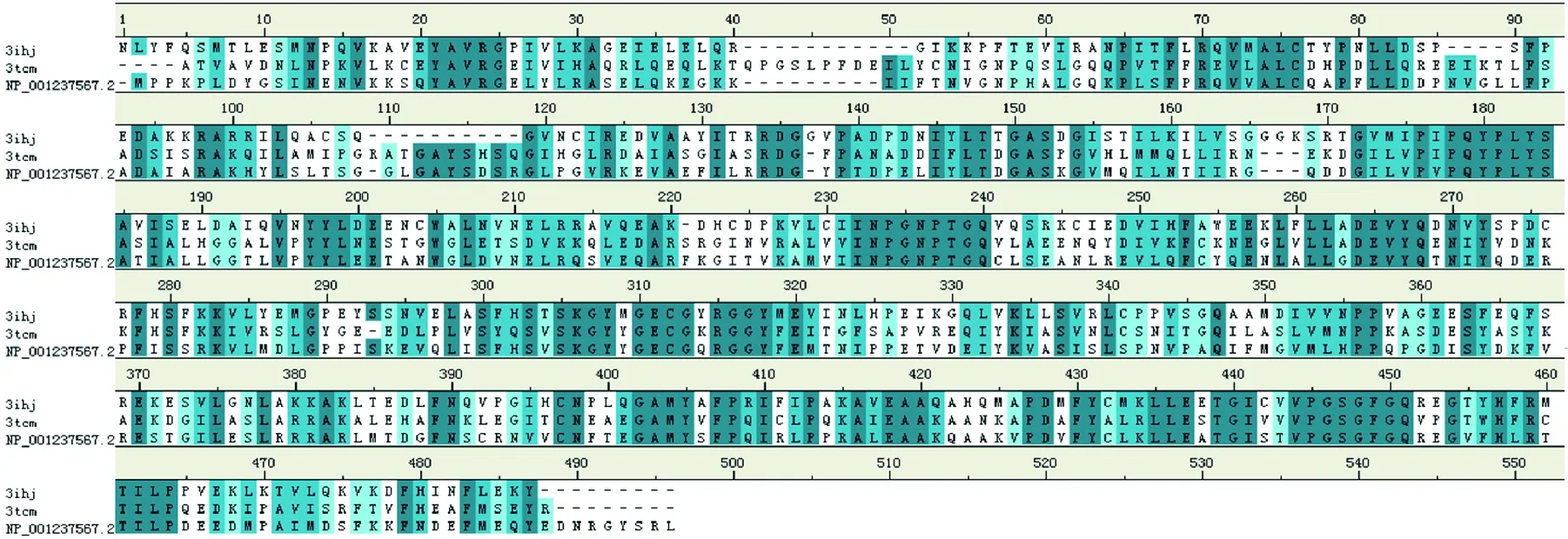
图 2 NP_001237567.2,3TCM和3IHJ序列对比图
1.4 GmGPT组氨酸标记模型的探究
在NP_001237567.2序列的C端加上6个组氨酸标签,利用Discovery Studio的Build Homology Models模块,以3TCM和3IHJ为模板构建大豆谷丙转氨酶模型,得到模型C,让模型C得到与A相同的loop修饰,然后将其置于0.20 mol/L、pH 7的KH2PO4-Na2HPO4模拟缓冲溶液中,依次采用Steepest Descent及Conjugate Gradient算法,分别固定蛋白质整体、蛋白质主链及全部柔性,对模型能量最小化,得到模型D[12]。探究模型D的C端组氨酸标签是否会进入蛋白质内部及组氨酸标签与活性位点的距离,从而判断组氨酸标记GmGPT的可行性。通过Discovery Studio的Calculation Protein Ionization and Residue pK模块,计算得到模型D的等电点,为后续表达和分离纯化GmGPT的工作进行理论准备。
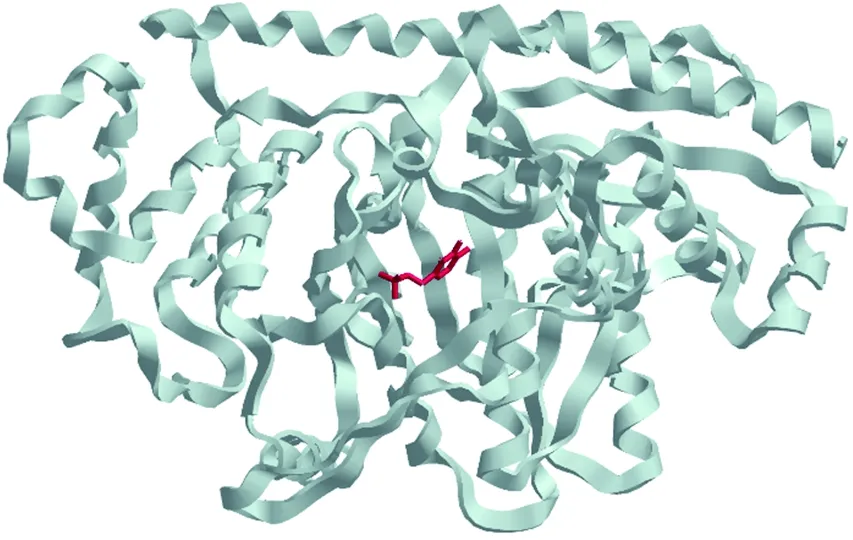
图为NP_001237567.2序列经同源建模所得到的单体模型A。图中蛋白质主链为灰白色;辅基PLP为樱桃红色
2 结果与分析
2.1 模型A结构及其合理性探究
通过同源建模和loop修饰得到模型,并将可能的辅基PLP的醛基与活性中心Lys291的ε-NH2以Schiff碱连接,得到切除了C端Asp474至Leu4818个残基的不匹配序列后的模型A(图3)。在UCLA的Saves平台验证后,模型的Verify Score为91.97%,Ramachandran Plot如图4。Ramachandran Plot显示模型中氨基酸残基93.7%位于最合适区,5.0%位于允许区,1.0%位于最大允许区,只有0.3%位于不允许区。上述结果显示出模型A质量较好。
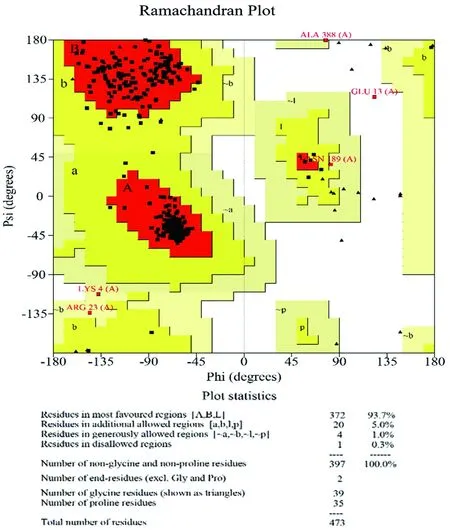
图4 大豆谷丙转氨酶模型A的Ramachandran Plot
2.2 模型B结构及其合理性探究
将模型A在上述溶液中进行分子动力学模拟后,得到6个不同的模型,这6个模型通过UCLA的Saves平台评估得到的Verify 评分、Errat评分和Prove评分如表2。可见当缓冲液浓度为0.2 mol/kg时,GmGPT模型的各项评分十分突出,其Verify 评分为97.67%,Errat评分为92.0430,Prove评分为0.0%,表明所建模型是合理的。其结构图如图5,中心活性位点结构图如图6。该模型有两个结构域,结构域I由Pro5到Phe70和Ile323到Glu473两端氨基酸序列组成,结构域II由Ala83到Ser322氨基酸序列组成,具体如图7。利用Discovery Studio的Dock Ligands (LibDock)模块,分别将谷氨酸、丙酮酸、α-酮戊二酸和丙氨酸与模型B进行刚性对接,对接均非常成功,结合位置均在Lys291-PLP附近。可见模型B对GPT的4种底物均具有较高的亲和性,且Lys291-PLP为酶的活性位点。
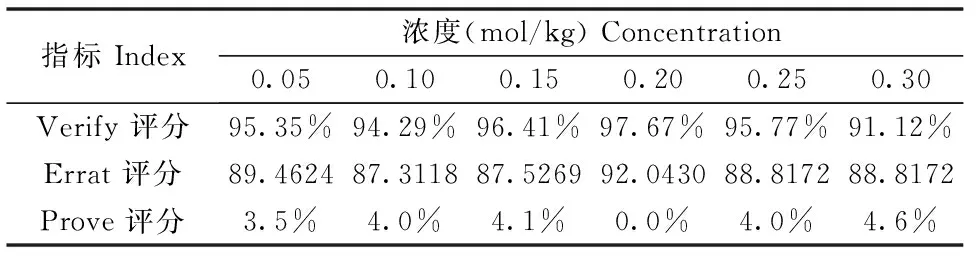
表2 不同浓度缓冲溶液模型的各项评分指标
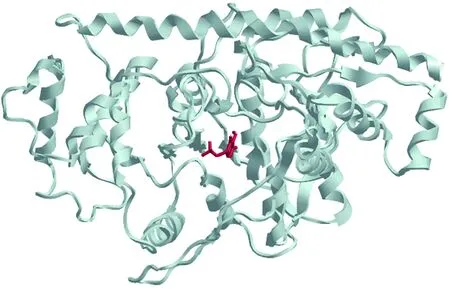
图为模型A在模拟缓冲溶液中进行能量最小化后所得到的模型B。图中蛋白质主链为灰白色;辅基PLP为樱桃红色
我们恢复了出发序列C端Asp474至Leu4818个残基的不匹配序列后,再于C端增加6个His残基作为组氨酸标签,得到表达序列。其同源建模后的模型为模型C,如图8-A。对其进行与A同等的loop修饰并在溶液状态按照与模型B同样的方法模拟后,得到模型D,如图8-B。模型D仅裸露出C端包含组氨酸标签的Asp474至His48714个残基在外侧,而且距离活性位点较远,可以忽略其对蛋白质结构的影响[13]。通过Discovery Studio的Calculation Protein Ionization and Residue pK模块,计算得到模型D的等电点为6.51。
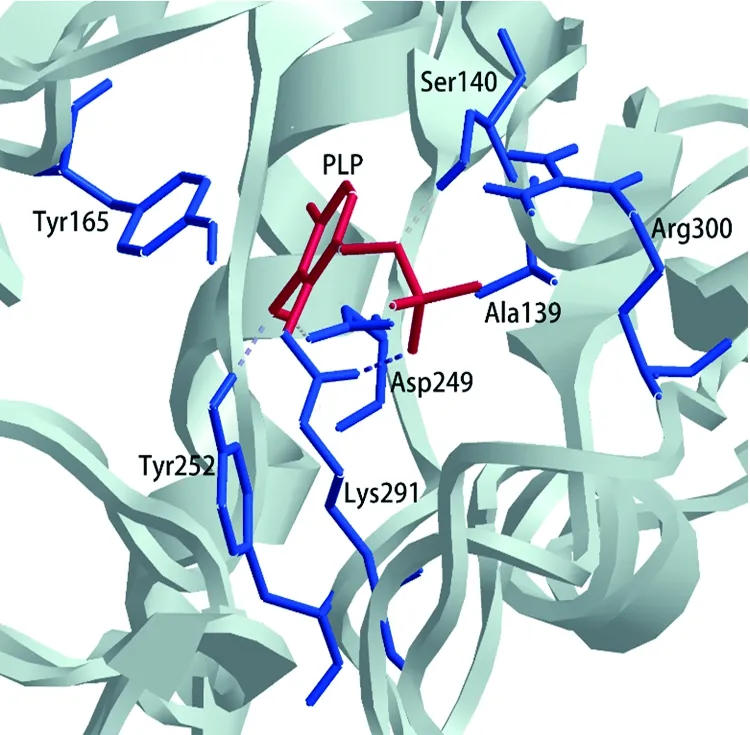
图为模型B活性位点与辅基PLP有相互作用的Ala139,Ser140,Tyr165,Asp249,Tyr252,Lys291,Arg300。图中蛋白质主链为灰白色;辅基PLP为樱桃红色;有相互作用的氨基酸为蓝色
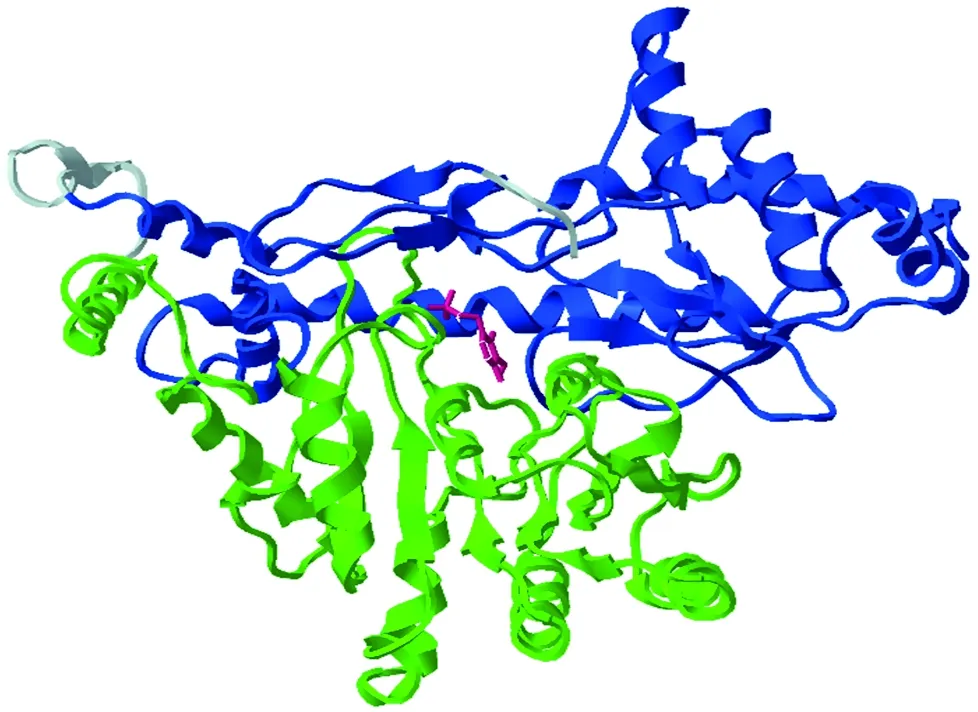
图为模型B的结构域。图中结构域I为蓝色;结构域II为绿色;辅基PLP为樱桃红色;其余氨基酸为灰白色
2.3 组氨酸标记模型的探究
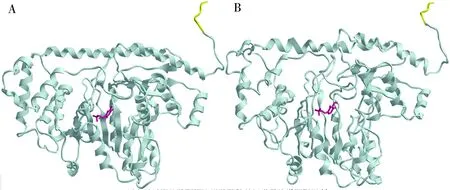
A为NP_001237567.2序列在C端添加组氨酸标签后,经同源建模所得到的单体模型C。B为模型C在模拟缓冲溶液中进行能量最小化后所得到的模型D。图中NP_001237567.2序列所编码的蛋白质主链为灰白色;组氨酸标签为黄色;辅基PLP为樱桃红
因而可通过亲和层析、分子凝胶过滤和离子交换柱层析纯化后,利用胰凝乳蛋白酶水解Tyr478和Ser479之间的肽键,得到除去标签且纯度较高的GmGPT。
3 讨论与结论
GPT在各种生物的代谢中起到重要的作用,而大豆中的GPT更是能在大豆内涝缺氧和缺氧后氧浓度恢复的情况下高效利用溶液中的氮,使植物组织保持活性[11]。对于蛋白质研究来说,其结构模型是活性和功能等研究工作的基础,而碍于解析晶体的技术原因,利用计算机同源建模成为一种经济而又具有一定准确性的方法[14]。但目前结构明确的植物GPT仅有来源于大麦(Hordeumvulgare)的GPT(PDB登录号3TCM)。
利用分子动力学模拟得到目标分子来观察其结构合理性可大大提高研究材料的可靠性。采用组氨酸标签可通过更改基因序列将带电序列加入到多肽链中,大大减轻了活性聚合的难度,提高了生物相容性和可降解性,因此组氨酸序列也常常和对环境敏感的嵌段活性聚合于某种肽链两端,形成A-C-B三嵌段聚合物,利用其在不同环境释放速度不同的特点制成缓释剂[15-16]。通过对三嵌段聚合物进行分子动力学模拟可了解其溶解度等相关的物理性质,同时也可得知组氨酸标签对肽段结构的影响,为进一步的实验研究筛选可执行方案,且此方法已在提纯蛋白质的研究中被使用[17]。
本研究中使用大豆谷丙转氨酶的可能序列(NP_001237567.2)作为靶序列,可以构建出与其他来源的GPT结构相似的模型,且该模型在模拟溶液中能够形成合理的构象,表现出与谷氨酸、丙酮酸、α-酮戊二酸和丙氨酸良好的对接能力,均证实了其作为大豆谷丙转氨酶的三维模型的合理性。对溶液态模型B的活性位点及结构域的分析将为继续深入研究大豆GPT乃至植物GPT的结构功能奠定前期工作基础。同时,该序列在C端连接组氨酸标签后,可能表达形成分子量约为54.57 ku、等电点约为6.51、组氨酸标签裸露且远离活性位点的单体蛋白质,证实了通过亲和色谱实现后续GmGPT的表达、纯化的可行性。
综上,GmGPT三维模型的构建对于丰富资源匮乏的植物GPT三维模型有着重要的意义,也能为大豆的氨基酸代谢的相关基础研究提供参考。
