代谢工程改造大肠杆菌合成L-组氨酸
2021-07-05李梦莹吕雪芹刘延峰李江华堵国成吴剑荣刘龙
李梦莹,吕雪芹,刘延峰,李江华,堵国成,吴剑荣,刘龙*
1(江南大学 未来食品科学中心,江苏 无锡,214122)2(糖化学与生物技术教育部重点实验室(江南大学),江苏 无锡,214122)
L-组氨酸,又名α-氨基-β-咪唑基丙酸,是分子中含有咪唑核的碱性氨基酸[1]。在营养学的范畴里,组氨酸被认为是婴幼儿必需的氨基酸[2]。L-组氨酸具有多种生理功能,对生长、组织修复以及治疗溃疡和胃酸过多等均具有重要的作用,还可以作为添加剂用于治疗过敏、风湿性关节炎以及贫血等疾病,因此,其被广泛应用于医药食品等行业[3]。
生产组氨酸的方法主要有蛋白水解法、化学合成法和微生物发酵法。其中水解蛋白质是组氨酸生产最为传统的方法[4],但是,这种方法主要取决于富含天然蛋白质的资源(如血粉或大豆)的可用性,很难满足人们对组氨酸日益增长的需求[5];而通过化学合成法生产组氨酸容易产生外消旋混合物,通常被认为是“非天然”化合物,难以获得食品药品监督管理局的批准,也较难被消费者接受[5];微生物发酵法是目前生产L-组氨酸的主流方法,因此L-组氨酸高产菌的选育成为当前的研究热点。常用于生产L-组氨酸的微生物主要包括大肠杆菌(Escherichiacoli,E.coli)、谷氨酸棒状杆菌(Corynebacteriumglutamicum,C.glutamicum)和粘质沙雷氏菌(Serratiamarcescens,S.marcescens)。不少研究者采用诱变结合组氨酸类似物筛选法选育L-组氨酸高产菌株。比如,KINO等[6]以具有1,2,4-三咪唑丙氨酸抗性的突变E.coliATCC21318为出发菌株,经诱变处理后获得1株组氨酸产量为14.2 g/L的菌株;以C.glutamicum为出发菌株,许益清等[7]通过定向进化选育出1株L-组氨酸为1~1.6 g/L的突变株;侯颖等[8]通过硫酸二乙酯(diethyl sulfate,DES)、亚硝基胍(nitroso guanidine,NTG)和紫外(ultra violet,UV)逐级诱变,获得1株高产L-组氨酸的S.marcescens突变菌株,L-组氨酸产量由最初的2.0 g/L提高到7.6 g/L。化学诱变等非理性设计虽然能筛选出高产菌株,但该方法具有很大的盲目性和随机性,往往很难获得稳定的高产菌株[7]。
理性改造手段由于具有目的性强、劳动强度低、效果显著、菌株稳定性好等优势,目前已经被广泛应用[3]。本研究首先对E.coliBL21(DE3)中L-组氨酸操纵子进行改造,解除L-组氨酸对于操纵子前导肽的调控;然后将来源于S.marcescensZJZ626[8]的ATP转磷酸核糖基酶编码基因hisG和核糖磷酸二磷酸激酶编码基因Prs利用CRISPR/Cas9技术整合至基因组,获得重组菌株B25,解除L-组氨酸对hisG的调控并增强前体物质5-磷酸核糖-1-焦磷酸 (5-phosphoribosy-l-pyrophosphate,PRPP)的合成;最终菌株B25中L-组氨酸的产量由最初的8.5 mg/L增加至5 574.63 mg/L。
1 材料与方法
1.1 宿主和质粒
本实验所有质粒的构建均在E.coliDH5α中进行,出发菌株为E.coliMG1655和E.coliBL21(DE3)。菌株E.coliDH5α、E.coliMG1655、E.coliBL21(DE3)、S.marcescensZJZ626、C.glutamicumATCC130132,质粒pET28a(+)均为本实验室保存,质粒pBSG11[9]为周哲敏教授赠送,菌株详情见表1,质粒详情见附表1(https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CAPJ&dbname=CAPJLAST&filename=SPFX20201221 005),引物详情见附表2。

表1 本文所用菌株Table 1 Strains used in this study
1.2 载体构建和菌株获取
1.2.1 载体构建和菌株获取
使用附表2中的引物,以E.coliBL21(DE3)、C.glutamicumATCC130132、S.marcescensZJZ626的基因组和pBSG11、pET28a质粒为模板,经PCR扩增分别获取hisG、hisG(C.glutamicum)、hisG(S.marcescens)序列和相应的线性化质粒,进一步通过Gibson组装获取重组质粒pBSG11-hisG和pET28a-hisG、pBSG11-hisG(C.glutamicum)和pET28a-hisG(C.glutamicum)、pBSG11-hisG(S.marcescens)和pET28a-hisG(S.marcescens)。采用类似的方法,获取重组质粒pBSG11-hisG(C.glutamicum)G233H/T235Q、pBSG11-hisG(C.glutamicum)G233H/T235Q/N215I、pBSG11-hisG(C.glutamicum)S143F/Δ209-281、pET28a-hisG(C.glutamicum)G233H/T235Q、pET28a-hisG(C.glutamicum)G233H/T23Q/N215I、pET28a-hisG(C.glutamicum)S143F/Δ209-281、pBSG11-hisG(S.marcescens)E271K、pET28-hisG(S.marcescens)E271K、pBSG11-hisGA10T、pBSG11-hisGC149Y、pBSG11-hisGA10T-C149Y-E271K、pET28a-hisGE271K、pET28a-hisGA10T、pET28a-hisGC149Y、pET28a-hisGA10T-C149Y-E271K、pBSG11-Prs、pBSG11-Prs(S.marcescens)。
1.2.2 基因组整合表达的菌株构建
以基因组motA位点整合hisG(S.marcescens)为例,首先以质粒pBSG11-hisG(S.marcescens)为模板扩增hisG(S.marcescens)基因,将Tac启动子通过上游引物引入到目的基因前,以E.coliBL21(DE3)为模板扩增上下游同源臂。采用融合PCR的方法[10]进行上下游同源臂及目的基因的融合,从而构建整合框。
选择整合位点motA,通过使用在线网站CHOPCHOP(https://chopchop.cbu.uib.no/)对sgRNA进行打分及排名,从而获得合适的sgRNA序列[11]。将位点motA的sgRNA设计为:5′-GCCGCAACAATACCAAACGC-3′,以实验室保存的pTarget质粒为模板,以pT-motA-F/R为引物进行反向PCR扩增,42 ℃热激90 s后,转化至E.coliDH5α感受态细胞中,单菌落测序正确后,获得质粒pTarget-motA。
将质粒pTarget-motA及整合框同时按照文献报道方法[12]转化至含有pCas质粒的B1中,对测序正确的菌株进行质粒pTarget-mota及pCas9的消除,获得重组菌株B16。采用上述类似的方法,获取重组菌株M1、M2、B1、B2、B17、B18、B24、B25。
1.3 摇瓶发酵方法
挑取单菌落接种于2 mL 液体LB培养基(酵母粉 5 g/L,胰蛋白胨 10 g/L,NaCl 10 g/L)中,37 ℃、220 r/min过夜培养。按4%(体积分数)将种子液接种于含有15 mL发酵培养基(葡萄糖 40 g/L,酵母粉 2 g/L,(NH4)2SO416 g/L,K2HPO4·3H2O 0.6 g/L,FeSO4·7H2O 0.005 g/L,MnSO4·5H2O 0.005 g/L,CaCO330 g/L)的250 mL摇瓶中,30 ℃、220 r/min培养72 h。
1.4 粗酶液制备方法
挑取单菌落接种于含有100 mg/L氨苄霉素的2 mL 液体LB培养基中,37 ℃、220 r/min过夜培养。按4%(体积分数)将种子液接种于含有25 mL TB培养基(酵母粉 24 g/L,胰蛋白胨 12 g/L,K2HPO412.54 g/L,KH2PO42.31 g/L)的250 mL摇瓶中,37 ℃、220 r/min培养至OD600为0.6~0.8,添加终浓度为0.1 mmol/L的异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG),16 ℃过夜诱导培养。将上述诱导结束后的菌液于4 ℃、7 000 r/min条件下离心10 min,弃上清液收集菌体,用250 mmol/L Tris-HCl(pH 8.1)洗涤2次,稀释菌体至OD600为8.0,冰水浴超声破碎后(功率300 W,工作4 s,间歇6 s,10 min),4 ℃、12 000 r/min离心30 min,收集上清液。
1.5 酶活力的测定
hisG酶活力的测定是基于反应产物磷酸核糖三磷酸腺苷(phosphoribosyl adenosine triphosphate,PR-ATP)和随后产生的磷酸核糖腺苷(phosphoribosyl adenosine monophosphate,PR-AMP)在290 nm处有较高的紫外吸收[13-15]。反应混合物(95 μL):79 μL反应溶液[Tris 6.07 g/L、KCl 5.59 g/L、MgCl20.48 g/L,另外可根据研究需求加入50~200 mmol/LL-组氨酸、2~16 mmol/L一磷酸腺苷(adenosine monophosphate,AMP)、1~5 mmol/L阿卡地新(AICAR),pH调至8.5]、1 μL 0.2 U/mL焦磷酸酶(pyrophosphatase,PPase),5 μL 0.2 U/mL ATP,10 μL 0.5 μg/μL粗酶液。将上述反应混合物置于96孔板中,30 ℃温浴5 min,加入5 μL 10 mmol/L PRPP,用5 μL的水替代PRPP用作空白对照。利用酶标仪测量该体系在290 nm、30 min内每30 s间隔的吸光度[16-17]。每分钟增加1吸收峰对应100 μL体系中形成0.091 9 μmol PR-ATP[16]。
1.6 检测方法
发酵液稀释至合适浓度的范围后,使用分光光度计测定其OD600值;采用SBA-40E系列生物传感分析仪测定发酵液中葡萄糖浓度;采用HPLC对发酵液中L-组氨酸浓度进行定量分析,以邻苯二甲醛进行发酵液柱前衍生,色谱柱为Agilent C18柱(250 mm×4.6 mm,5 μm),紫外检测器的检测波长为338 nm,柱温40 ℃,进样量1 μL。具体的梯度洗脱程序如表2所示。

表2 本文梯度洗脱程序Table 2 Gradient eluting procedure used in this study
2 结果与分析
2.1 hisLGDCBHAFI操纵子前导区改造对L-组氨酸合成和细胞生长的影响
E.coli中合成L-组氨酸的途径酶编码基因组成1个 操纵子,基因顺序为hisLGDCBHAFI。该操纵子受L-组氨酸调控:当L-组氨酸浓度高时,核糖体快速跳过操纵子中hisL编码的前导肽,在hisL和hisG之间形成终止子,从而阻止转录的进行;当L-组氨酸浓度低时,核糖体与hisL基因结合,进行前导肽的合成[3](图1-a)。为了解除L-组氨酸对操纵子的调控,用人工的开放阅读框(hisL′-λattB′-trpE)[18-19]分别替换E.coliBL21 (DE3)、E.coliMG1655中的L-组氨酸操纵子的前导区(图1-b),得到菌株B1、M1。已知PurR是E.coli中响应嘌呤和嘧啶代谢的调节子,通过影响代谢前体PRPP和代谢中间物AICAR的合成量调控组氨酸代谢[18,20],因而,为了解除PurR对L-组氨酸代谢的影响,敲除菌株B1、M1中的PurR基因[10,21],依次得到菌株B2、M2。
ATP转磷酸核糖激酶(E.coli中由hisG基因编码)的催化是L-组氨酸合成通路中的第一步反应,也是L-组氨酸合成的限速步骤[16]。有研究发现,将E.coli中ATP转磷酸核糖激酶的第271位谷氨酸突变为赖氨酸,可以在一定程度上解除L-组氨酸的反馈抑制[18]。因而,将该突变基因插入pBSG11质粒中,获取重组质粒pBSG11-hisGE271K,并将其分别转化至菌株E.coliBL21(DE3)、E.coliMG1655、B1、M1、B2、M2 中,得到菌株B3、M3、B4、M4、B5、M5。摇瓶发酵72 h后,结果如图1-c所示,菌株B4的L-组氨酸产量最高,为872.50 mg/L,是对照菌株B3产量的3.68倍;同时,B4菌株的OD600是B3的0.48倍。上述实验结果说明,操纵子前导区的改造对L-组氨酸的合成有较明显的促进效果,因此,接下来的研究中选择菌株B1为主要研究对象。
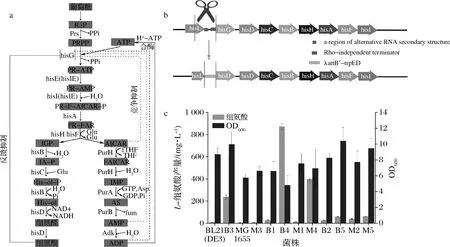
a-E.coli L-组氨酸合成路径;b-替换L-组氨酸操纵子前导肽示意图;c-不同菌株的L-组氨酸产量和OD600分析图1 E.coli的L-组氨酸合成途径及hisLGDCBHAFI操纵子前导区改造对L-组氨酸合成及细胞生长的影响Fig.1 Synthesis pathway of L-histidine in E.coli and effects of hisLGDCBHAFI operon′s leading area modification on L-histidine synthesis and cell growth
2.2 ATP转磷酸核糖激酶hisG改造对L-组氨酸合成的影响
2.2.1 在B1中表达C.glutamicum来源hisG基因验证其对L-组氨酸合成的影响
已有研究报道C.glutamicumATCC13032来源的hisG基因可以通过定点突变在一定程度上解除L-组氨酸的反馈抑制,进而提高C.glutamicum中L-组氨酸的产量[16,21]。因此将C.glutamicumATCC13032来源的天然hisG基因(C1)及其突变基因hisGG233H/T235Q(C2)、hisGG233H/T235Q /N215I(C3)、hisGS143F/Δ209-281(C4)插入pBSG11质粒中,获取重组质粒并将其分别转化至菌株B1,得到菌株B7、B8、B9、B10。同时,使用载体pBSG11过表达E.coli天然hisG基因(EG),获得重组质粒pBSG11-hisG,将其转化至菌株B1中,获得对照菌株B6。72 h摇瓶发酵结果显示,菌株B8的L-组氨酸产量为1 070.79 mg/L,OD600为5.82(图2-a)。
Ecoli天然hisG基因编码的ATP转磷酸核糖激酶,受到L-组氨酸的反馈抑制[22],AMP[23]、AICAR[24]的竞争性抑制。为了验证上述5种hisG基因所编码的ATP转磷酸核糖激酶在不同浓度抑制剂下的酶活,分别将EG、C1、C2、C3、C4插入表达载体pET28a,获得重组质粒pET28a-hisG、pET28a-hisG(C.glutamicum)G233H/T235Q、pET28a-hisG(C.glutamicum)G233H/T235Q/N215I、pET28a-hisG(C.glutamicum)S143F/Δ209-281,转化至E.coliBL21(DE3)中,按照1.5中方法收集胞内上清液并利用SDS-PAGE检测蛋白的表达情况。如图2-b所示,5种hisG蛋白均成功表达,其中EG、C1、C2、C3表达量均高于C4。在反应体系中组氨酸浓度达到200 mmol/L时,C2、C3、C4仍能保持>60%的相对酶活力(图2-c)。在AICAR浓度为0~3 mmol/L时,仅C2酶活力高于对照EG及C1;AICAR浓度达到5 mmol/L时,C1,C2、C3、C4的相对酶活为EG的1.15~1.45倍(图2-d)。上述5种酶,在反应体系中含有2 mmol/L AMP时,酶活力均下降至20%以下(图2-e)。
2.2.2 在B1中表达S.marcescens来源hisG基因验证其对L-组氨酸合成的影响
实验室存有1株经过诱变筛选后具有组氨酸结构类似物(3-氨基-1,2,4-三氮唑、6-巯基嘌呤、组氨酸甲酯、2-硫尿嘧啶、D-组氨酸)抗性的S.marcescensZJZ625[8],使用附表2中引物克隆其hisG基因(S1)并进行测序(附表3),同时对基因S1的271位氨基酸通过PCR进行定点突变,得到hisG(S.marcescens)E271K(S2)。在菌株B1中,使用pBSG11载体过表达上述基因S1、S2,得到菌株B11、B12。72 h摇瓶发酵结果显示(图3-a),菌株B11的L-组氨酸产量达到1 480.42 mg/L,为对照菌株B6的10.86倍,OD600下降至5.57。271位氨基酸的突变虽然未对OD600产生明显影响,但导致了产量的下降(894.80 mg/L)。

a-过表达C.glutamicum来源hisG菌株的L-组氨酸产量及OD600;b-C.glutamicum来源突变hisG的SDS-PAGE图 [1-对照,2-pET28a-hisG,3-pET28a-hisG(C.glutamicum),4-pET28a-hisG(C.glutamicum)G233H-T235Q,5-pET28a-hisG(C.glutamicum)G233H-T235Q-215I, 6-pET28a-hisG(C.glutamicum)S143F/Δ209-281]; c-hisG(C.glutamicum)在不同组氨酸浓度下的相对酶活力;d-hisG(C.glutamicum) 在不同AICAR浓度下的相对酶活力;e-hisG(C.glutamicum)在不同AMP浓度下的相对酶活力图2 表达C.glutamicum来源hisG对L-组氨酸合成的影响Fig.2 Effect of expression of hisG from C.glutamicum on L-histidine synthesis
分别将S1、S2基因片段插入pET28a载体,获得重组载体pET28a-hisG(S.marcescens)、pET28a-hisG(S.marcescens)E271K,将其转化至E.coliBL21(DE3),低温诱导蛋白表达后,分析胞内上清液(图3-b)。在反应体系中含有200 mmol/LL-组氨酸时,S1仍能保持64.57%的相对酶活力(图3-c);AICAR浓度在0~5 mmol/L,EG、S1、S2的酶活无明显差异(图3-d);在AMP浓度为2 mmol/L时,S1仍能保持79.77%的相对酶活力(图3-e)。
2.2.3 改造E.coli的野生型hisG基因并验证其对L-组氨酸合成的影响
菌株S.marcescensZJZ625诱变前的基因序列未知,为了探究导致L-组氨酸产量提高的有效突变位点,将E.coliBL21(DE3)、S.marcescensSMDB11、S.marcescensZJZ625的hisG基因的氨基酸序列进行比对(图4-a),选择S.marcescensZJZ625的hisG基因中与E.coliBL21(DE3)、S.marcescensSMDB11均不同的氨基酸位点,即为第10位与第149位氨基酸作为目标位点,同时由于已有研究报道271位氨基酸也为有效位点[18],将E.coli天然hisG的第10、149、271位氨基酸分别进行突变以及3个位点同时突变,得到突变基因hisGA10T(E1)、hisGC149Y(E2)、hisGE271K(E3)、hisGA10T/C149Y/E271K(E4),将其分别插入PBSG11载体,转化至菌株B1,获得菌株B13、B14、B4、B15。摇瓶发酵72 h后,如图4-b所示,菌株B15的L-组氨酸产量最高(1 148.923 mg/L),OD600为6.91。在E.coliBL21(DE3)中,使用pET28a载体表达E1、E2、E3、E4基因,低温诱导E1、E2、E3、E4蛋白表达后,分析胞内上清液(图4-c)。4种突变基因均在一定程度上解除了L-组氨酸的反馈抑制,在反应体系中含有200 mmol/LL-组氨酸时,仍能保持60%以上的酶活力(图4-d),但E4在4 mmol/L AICAR浓度下就失去酶活性 (图4-e),同时E4在反应体系中含有12 mmol/L AMP时仍能保持30%以上的酶活力(图4-f)。

a-过表达S.marcescens来源hisG菌株的L-组氨酸产量及OD600;b-S.marcescens来源突变hisG的SDS-PAGE图 [1-对照,2-pET28a-hisG,3-pET28a-hisG(S.marcescens),4-pET28a-hisG(S.marcescens)E271K]; c-hisG(S.marcescens)在不同组氨酸浓度下的相对酶活力;d-hisG(S.marcescens)在不同AICAR浓度下的相对酶活力;e-hisG(S.marcescens) 在不同AMP浓度下的相对酶活力图3 表达S.marcescens来源hisG对L-组氨酸合成的影响Fig.3 Effect of expression of hisG from S.marcescens on L-histidine synthesis
2.3 基因组整合hisG基因对L-组氨酸合成和细胞生长的影响
基于上述菌株B11的L-组氨酸产量最高,并在200 mmol/LL-组氨酸、5 mmol/L AICAR、2 mmol/L AMP浓度下均能保持较高酶活力。通过Crisper-Cas9技术在菌株B1的mota[25]、chew[25]、poxb[26]位点依次进行了hisG(S.marcescens)基因的整合,获得了菌株B16、B17、B18。如图5所示,菌株B17产量为861.78 mg/L,为菌株B16的1.31倍;但菌株B18产量提高不明显,OD600值也未发生明显变化。因此,推测前体物质供应不足限制了L-组氨酸的生产。

图5 基因组整合S.marcescens来源hisG对 L-组氨酸合成及细胞生长的影响Fig.5 Effects of genomic integration of hisG from S.marcescens on L-histidine synthesis and cell growth
2.4 前体供应关键基因Prs对L-组氨酸合成的影响
核酮糖-5-磷酸(ribulose-5-phosphate,R5P)经过磷酸核糖焦磷酸合成酶(phosphoribosyl pyrophosphate synthetase,Prs)的催化生成L-组氨酸合成的前体物质PRPP[21]。为了增强前体物质PRPP的供应,在菌株B16、B17、B18中使用pBSG11载体过表达Prs基因,获得菌株B19、B20、B21。摇瓶发酵72 h后,菌株B21产量达到2 119.28 mg/L,OD600达到9.83(图6-a)。
为了验证过表达S.marcescensZJZ625来源的Prs基因(附表4)对L-组氨酸合成的影响,构建重组质粒PBSG11-Prs(S.marcescens),转化至产量较高的菌株B20和B21,得到菌株B22、B23。72 h 摇瓶发酵结果显示,过表达Prs(S.marcescens)比过表达E.coli的野生型Prs提高L-组氨酸产量效果更好,其中菌株B23的L-组氨酸产量提高至2 778.26 mg/L(图6-b)。
将Prs(S.marcescens)在菌株B18的ylbG[27]、recA[27]位点依次进行基因组整合,得到菌株B24、B25。菌株B25摇瓶发酵72 h后,产量达到3 898.06 mg/L,是菌株B23的1.40倍(图6-c)。若继续延长发酵时间至96 h,菌株B24(OD600=6.45)、菌株B25(OD600=4.81)的L-组氨酸产量分别提高至5 040.69、5 574.63 mg/L,菌株B25 96 h消耗葡萄糖总量为72 h消耗葡萄糖量的1.24倍(图6-d)。

a-E. coli BL21 (DE3)、S. marcescens ZJZ626和S. marcescens SMD11的hisG氨基酸序列对比图;b-过表达E. coli 来源hisG菌株的L-组氨酸产量及OD600;c-E. coli来源突变hisG的SDS-PAGE图(1-对照,2-pET28a-hisG, 3-pET28a-hisGA10T,4-pET28a-hisGC149Y,5-pET28a-hisGE271K,6-pET28a-hisGA10T/C149Y/E271K); d-hisG在不同组氨酸浓度下的相对酶活力;e-hisG在不同AICAR浓度下的相对酶活力;f-hisG在不同AMP浓度下的相对酶活力图4 改造E.coli来源hisG对L-组氨酸合成的影响Fig.4 Effect of expression of hisG from E.coli on L-histidine synthesis
3 结论
L-组氨酸合成路径较长,通过诱变单一地选育结构类似物抗性突变株并不会完全打通菌体内合成L-组氨酸的代谢途径。本研究利用代谢工程思路对E.coliL-组氨酸合成的代谢途径进行了改造,构建无质粒高产大肠杆菌。首先通过CRISPR/Cas9技术对组氨酸操纵子前导肽进行替换,解除了L-组氨酸对操纵子转录的调控。组氨酸合成途径中的第一个酶(hisG)会受到L-组氨酸的反馈抑制以及AMP、AICAR的竞争性抑制,因此我们分析了表达C.glutamicum、S.marcescens、E.coli来源的不同hisG对L-组氨酸产量及细胞生长的影响,同时分析了hisG基因所编码的ATP转磷酸核糖激酶在不同浓度L-组氨酸、AMP、AICAR时的酶活力,最终选择了对提高L-组氨酸产量效果最好,同时对3种抑制物均有较高耐受浓度的hisG(S.marcescens)进行基因组整合,整合3个拷贝hisG(S.marcescens)的菌株B18L-组氨酸产量提高至905.32 mg/L。在实验过程中发现,菌株B18相对于基因组整合2个拷贝hisG(S.marcescens)的菌株B17产量提升并不明显,推测前体物质供应不足限制了L-组氨酸的生成。所以尝试过表达控制前体PRPP合成的基因Prs,产量提高至2 119.28 mg/L。接着又比较了S.marcescens来源的Prs对L-组氨酸产

a-在不同菌株中表达PBSG11-Prs质粒对L-组氨酸生产及细胞生长的影响;b-表达不同来源Prs对L-组氨酸生产及细胞生长的影响; c-不同拷贝数的Prs(S.marcescens)对L-组氨酸生产及细胞生长的影响;d-不同发酵时间对L-组氨酸生产、耗糖量及细胞生长的影响图6 提高PRPP的合成对L-组氨酸产量及OD600的影响Fig.6 Effect of increasing PRPP synthesis on L-histidine titer and OD600
量的影响,发现其产量为过表达E.coli天然Prs菌株的1.31倍,将Prs(S.marcescens)进行2个拷贝的基因组整合后,获得重组菌株B25,摇瓶发酵72 hL-组氨酸产量提高至3 898.06 mg/L,96 hL-组氨酸产量达到5 574.63 mg/L。该研究为实现更经济的L-组氨酸工业生产奠定了基础。
