磷改性对NiMo/γ-Al2O3硫化物催化剂硫醚化性能的影响
2020-08-15李风旭李明丰陈吉祥
李风旭,李明丰,褚 阳,陈吉祥*
(1.天津大学化工学院,催化科学与工程系,天津市应用催化科学与工程重点实验室,天津 300350; 2.中国石化石油化工科学研究院,北京 100083)
汽油中含硫化合物燃烧会产生大气污染物SO2,其中FCC汽油含有绝大部分硫化物。传统加氢脱硫工艺虽然可以降低汽油中硫含量,但同时也会使汽油中的烯烃加氢饱和,既降低了汽油辛烷值也增加了耗氢量。此外,FCC汽油中少量的二烯烃在催化剂表面极易发生聚合、积炭而导致催化剂失活。为此,基于硫醚化反应与馏分切割而获得超低硫含量催化裂化汽油的Prime-G+工艺已广泛应用。
目前,硫醚化催化剂的研究相对较少。已报道的硫醚化催化剂包括金属、金属硫化物、过渡金属磷化物以及固体酸。过渡金属硫化物催化剂在含硫条件下具有良好的稳定性,广泛应用于油品加氢脱硫及脱氮过程。因而,过渡金属硫化物(尤其是硫化的Ni-Mo/γ-Al2O3)为研究和应用较多的硫醚化催化剂。肖招金等[1-3]发现,硫化后Ni/Al2O3催化剂具有良好的硫醚化性能。申志兵等[4-6]采用金属Mo改性,显著提高了Ni/Al2O3催化剂硫醚化性能。通常认为Ni-Mo/γ-Al2O3硫化物催化剂上硫醚化反应与二烯烃选择性加氢反应的活性中心均为Ni-Mo-S活性相[5,7],因此硫醚化与选择性加氢为竞争反应。另有研究表明,催化剂表面的酸性位为硫醚化反应活性中心[8]。本课题组前期工作[9-10]也表明过渡金属磷化物表面P—OH基团可促进硫醚化反应。我们认为提高Ni-Mo/γ-Al2O3硫化物催化剂表面酸性是改善其硫醚化性能一种手段。因此,本研究采用磷对Ni-Mo/γ-Al2O3硫化物催化剂进行改性,旨在提高Ni-Mo/γ-Al2O3催化剂硫醚化性能。由于浸渍次序影响各组分与载体之间的相互作用[11],研究了磷物种及Ni-Mo金属组分浸渍次序对催化剂结构和性能的影响。
1 实验部分
1.1 催化剂制备
采用等体积共浸渍及分步浸渍法制备催化剂氧化态前驱体。配制(NH3)6Mo7O24·4H2O、Ni(NO3)2·6H2O及NH4H2PO4混合溶液,等体积浸渍γ-Al2O3载体,经120℃干燥12 h及550 ℃焙烧4 h获得共浸渍制备催化剂前驱体。分别配制(NH3)6Mo7O24·4H2O与Ni(NO3)2·6H2O的混合溶液及NH4H2PO4溶液,先将(NH3)6Mo7O24·4H2O与Ni(NO3)2·6H2O混合溶液或NH4H2PO4溶液等体积浸渍γ-Al2O3载体,经120℃干燥12 h及550 ℃焙烧4 h得到负载Ni、Mo金属组分或负载P组分的样品,所得到样品再分别等体积浸渍NH4H2PO4溶液或(NH3)6Mo7O24·4H2O与Ni(NO3)2·6H2O混合溶液,再经120℃干燥12 h及550 ℃焙烧4 h获得先负载金属组分后负载P组分及先负载P组分后负载金属组分的催化剂前驱体。此外,采用(NH3)6Mo7O24·4H2O、Ni(NO3)2·6H2O混合溶液等体积浸渍γ-Al2O3载体,经120℃干燥12 h及550 ℃焙烧4 h获得未改性催化剂前驱体。
以质量分数2%CS2/环己烷溶液为硫化剂,在H2压力1.5 MPa、氢油体积比300、质量空速为2.5 h-1条件下采用程序升温对催化剂氧化态前驱体进行硫化:以2 ℃·min-1升温速率由室温升至150 ℃,通入硫化剂浸润催化剂1 h,然后继续升温,在250 ℃和290 ℃各保持2 h。硫化结束后,待降至反应温度,降至常压用H2(320 mL·min-1)吹扫1 h。
所制备催化剂中Ni、Mo及P质量分数分别为15%、5%及3%。未采用P改性的催化剂标记为NiMo;共浸渍法制备的催化剂标记为NiMoP;先浸渍金属组分后浸渍P组分的催化剂标记为NiMo-P;先浸渍P组分后浸渍金属组分催化剂标记为P-NiMo。
1.2 催化剂表征
采用X'Pert Pro粉末衍射仪进行XRD测试,Cu Kα(λ=0.154 18 nm)。
采用JEM-2100F场发射电子显微镜进行HRTEM表征。
采用NH3-TPD程序升温脱附法及CO化学吸附法表征催化剂表面酸性及金属位密度。
1.3 催化剂活性评价
采用不锈钢固定床反应器(内径12 mm)评价催化剂硫醚化性能。以质量分数0.4%异戊二烯和0.028%正丁硫醇(对应S含量为0.01%)的环己烷溶液为模拟油。催化剂装填量为1.0 g。在反应温度120 ℃、H2压力1.5 MPa、模拟油质量空速(WHSV)8 h-1以及H2/模拟油体积比为50条件下评价催化剂性能。采用北京分析仪器厂SP-3420型气相色谱仪对液相产物进行定量分析,FID检测器,SE-30毛细管柱(50 m×0.32 mm×3.0 μm),正辛烷为内标物。
2 结果与讨论
2.1 表征结果
2.1.1 H2-TPR
浸渍次序影响负载组分与载体γ-Al2O3之间的相互作用,从而影响负载组分分散度及硫化程度。采用H2-TPR对催化剂氧化态前驱体进行表征以获得催化剂前驱体中负载组分与载体相互作用的信息。

由图1还可知,与NiMo前驱体相比,NiMoP及NiMo-P前驱体在(300~600) ℃间主还原峰向高温方向偏移,与磷物种与金属组分产生相互作用抑制了Mo6+→Mo4+及Ni2+→Ni0的还原有关。NiMo、NiMoP及NiMo-P前驱体具均有3个还原峰(分别位于为435 ℃、520 ℃ 和730 ℃左右),而P-NiMo前驱体仅在(300~600) ℃出现一个明显的还原峰(峰顶温度约435 ℃),表明P-NiMo前驱体中金属物种易于还原且存在环境比较相近。在制备P-NiMo前驱体时,先浸渍的P物种在焙烧过程中易与载体γ-Al2O3发生相互作用形成表面AlPO4,减弱了后浸渍的金属组分与载体的相互作用,一方面降低了金属组分的还原温度[12],另一方面使负载金属组分具有相近的存在环境。总之,所制备的P改性前后的催化剂前驱体中,P-NiMo催化剂前驱体最容易被还原。

图1 催化剂氧化态前驱体的H2-TPR谱图Figure 1 H2-TPR profiles of catalyst precursors in oxidation state
2.1.2 XRD
图2为各催化剂氧化态前驱体XRD图。由图2可知,除载体γ-Al2O3外,各个催化剂衍射图中只发现了NiO的衍射峰(2θ=37.2°、43.3°、62.9°、75.4°与79.4°,PDF#47-1049);未检测到Mo与P物种,与其含量低、分散度高有关。与其他催化剂氧化物前驱体相比,P-NiMo催化剂前驱体具有较强的NiO衍射峰,与先浸渍P抑制了金属组分与载体Al2O3之间相互作用有关,导致了NiO晶粒尺寸增大,与H2-TPR结果一致。共浸渍法制备的NiMoP催化剂前驱体的NiO衍射峰较弱,即NiMoP催化剂前驱体中金属组分分散度较好。

图2 催化剂氧化态前驱体的XRD图Figure 2 XRD patterns of catalyst precursors in oxidation state
图3为氧化物前驱体硫化后制备的硫化物催化剂的XRD图。由图3可知,各硫化物催化剂XRD图中均存在较弱的Ni3S2衍射峰(2θ=29.7°与49.4°,PDF#27-0341),其中P-NiMo催化剂的Ni3S2衍射峰最为明显,与其前驱体中金属组分与载体相互作用较弱有关。硫化物催化剂中也未发现与Mo与P组分有关的衍射峰。
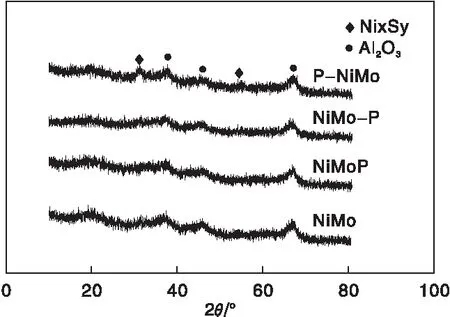
图3 硫化物催化剂的XRD图Figure 3 XRD patterns of sulfide catalysts
2.1.3 TEM
为进一步确认硫化物催化剂中是否有MoS2形成,对NiMo及NiMo-P硫化物催化剂进行TEM表征,结果如图4所示。由图4可知,NiMo及NiMo-P催化剂中均发现了Ni3S2晶粒,但仍未发现层状MoS2,可能与其含量较低有关。
2.1.4 NH3-TPD
图5为硫化物催化剂的NH3-TPD曲线,表1为硫化物催化剂的相对酸量。由图5可知,各硫化物催化剂在200 ℃左右均存在一个较大的脱附峰,与金属硫化物表面吸附NH3的脱附有关;此外,450 ℃较小脱附峰可能与未被硫化还原的金属离子上NH3的脱附有关[13-14]。在(300~400)℃间NiMo催化剂上未出现脱附峰,但P改性硫化物催化剂上出现了脱附峰,可能与NH3吸附在催化剂表面的P--OH有关。与NiMo硫化物催化剂相比,P改性硫化物催化剂具有较大的NH3吸附峰,即P改性提高了NiMo催化剂的酸量。原因包括:1)P组分与载体γ-Al2O3发生相互作用会形成AlPO4,其中Al-O--P-OH基团为B酸中心[15];2)未与载体发生相互作用的P物种以磷酸根形式存在时也可提供P--OH基团作为B酸中心。另外,由表1可知,NiMo-P与P-NiMo催化剂酸量高于NiMoP。

图5 硫化物催化剂的NH3-TPD曲线Figure 5 NH3-TPD profiles of sulfide catalysts
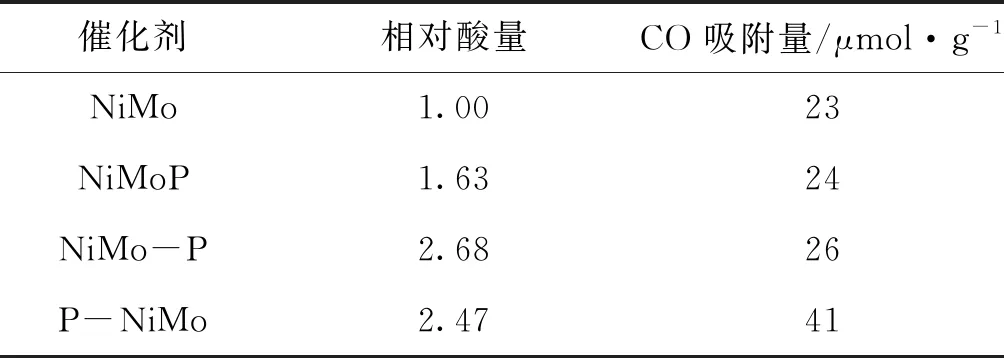
表1 硫化物催化剂的相对酸量与CO化学吸附量
2.1.5 CO化学吸附
采用CO化学吸附方法测定硫化物催化剂表面金属位密度,结果如表1所示。由表1可知,硫化物催化剂上CO化学吸附量由高到低的顺序为P-NiMo>NiMo-P>NiMoP>NiMo。可见,P改性不同程度提高了硫化物催化剂CO化学吸附量,与P改性降低了金属活性组分与载体γ-Al2O3间相互作用有关,从而利于金属物种转化为硫化物并暴露更多的金属位。其中,P-NiMo的CO化学吸附量最大。
2.2 硫醚化性能
以正丁硫醇转化率(XB)、C4组分选择性(SC4)、异戊二烯转化率(XIso)、C5组分总选择性(St-C5)及C5单烯烃选择性(SOle)表示催化剂性能。
2.2.1 正丁硫醇转化率及C4组分选择性
不同硫化物催化剂上正丁硫醇转化率如图6所示。由图6可知,各催化剂上XB由高到低次序如下:P-NiMo>NiMo-P>NiMo≈NiMoP。P-NiMo催化剂上XB最高,反应12 h后稳定于约93%。NiMo与NiMoP催化剂上XB相近,低于其他催化剂。NiMo-P催化剂上XB(反应12 h后约为74.6%)介于NiMoP与P-NiMo催化剂之间。

图6 不同硫化物催化剂上正丁硫醇转化率Figure 6 Conversion of n-butanethiol on different sulfide catalysts
不同硫化物催化剂上C4烃选择性如图7所示。由图7可知,随着反应的进行,NiMo与NiMoP催化剂上SC4均迅速增加;反应第3 h时SC4约为1%左右,反应至第15 h时增加至20%以上。然而,随反应进行,NiMo-P催化剂与P-NiMo催化剂上SC4相近且增加不明显(2.3~4.6%)。

图7 不同硫化物催化剂上C4烃选择性Figure 7 Selectivity of C4 hydrocarbons on different sulfide catalysts
如上所述,P-NiMo催化剂具有较高XB与较低SC4,表明其具有较好的硫醚化性能。先浸渍P组分使得P-NiMo催化剂氧化物前驱体更易被硫化、催化剂表面暴露较多的活性中心。并且,P物种与载体表面的Al-OH相互作用生成具有B酸特性的Al-O-P-OH基团,而P--OH基团有利于硫醚化反应进行。
2.2.2 异戊二烯转化率及C5组分选择性
图 8为不同硫化物催化剂上异戊二烯转化率。由图8可知,各催化剂上XIso由高到低次序为NiMoP>NiMo>NiMo-P>P-NiMo。其中,NiMoP催化剂上XIso最高(97.3 %),而P-NiMo上XIso最低(仅为67 %)。
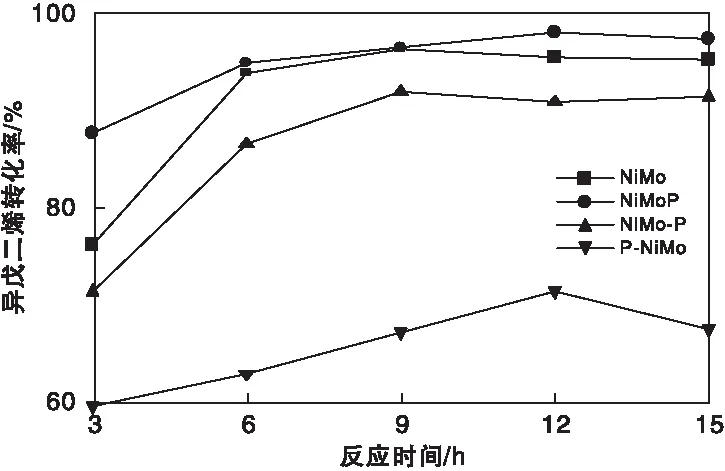
图8 不同硫化物催化剂上异戊二烯转化率Figure 8 Conversion of isoprene on sulfide catalysts
浸渍顺序对C5组分和单烯烃组分选择性的影响如图9所示。由图9可知,各催化剂上SC5与SOle均非常接近,表明各催化剂均具有良好的选择加氢性能。与NiMo催化剂相比,P改性催化剂上St-C5较低,与P组分加入提高了催化剂酸性、促进烯烃聚合生成高聚物有关。P改性催化剂中,P-NiMo酸量较大,其上St-C5最低(仅约70 %)。此外,NiMo-P催化剂的St-C5(82.4 %)略低于NiMoP催化剂(85.7%)。

图9 浸渍顺序对催化剂C5组分(实线)与单烯烃组分 (虚线)选择性的影响Figure 9 Effect of impregnation sequence on the selectivity to C5 components (solid line) and monoolefin component (dash line) on the catalysts
综合考虑异戊二烯转化率及产物选择性,各催化剂异戊二烯选择加氢性能由高到低顺序如下:NiMo>NiMo-P≈NiMoP>P-NiMo。
考虑到NiMo-P上正丁硫醇转化率仅低于P-NiMo,而NiMo-P催化剂的异戊二烯选择加氢性能仅低于NiMo催化剂,NiMo-P催化剂性能相对较佳。
3 结 论
(1) P物种引入抑制了Ni-Mo与载体γ-Al2O3的相互作用,促进了金属组分硫化、提高硫化物催化剂表面暴露金属位,其中先浸渍P后浸渍Ni-Mo所制备的催化剂尤为明显。所制备的硫化物中形成了Ni2S3物相,未形成MoS2物相。P组分的加入提高了NiMo/γ-Al2O3催化剂的酸量。
(2) 先浸渍及后浸渍P组分均提高了催化剂硫醚化性能,其中先浸渍P物种制备的P-NiMo催化剂性能较佳。然而,P物种和金属组分共浸渍制备NiMoP更利于异戊二烯转化。由于催化剂酸量增加,P改性提高了催化剂的烯烃聚合活性。P-NiMo催化剂异戊二烯选择加氢性能最低。
(3) 综合考虑,先浸渍金属组分后浸渍P组分制备的NiMo-P催化剂性能相对较好。
