磷酸吡哆醛的合成工艺
2020-08-13宣良明冯权武尹传奇
宣良明,胡 旭,冯权武,卢 伟,尹传奇*
1.武汉工程大学化学与环境工程学院,湖北 武汉430205;
2.湖北惠生药业有限公司,湖北 咸宁437000
磷酸吡哆醛,化学名称为2-甲基-3-羟基-5-(磷酰氧基)甲基-4-吡啶甲醛,是一种重要的维生素B6族化合物[1-2],不仅是氨基酸代谢中的转氨酶及脱羧酶的辅酶[3],还能促进谷氨酸脱羧[4],增进γ-氨基丁酸生成[5]。磷酸吡哆醛还大量应用在合成β-羰基酰胺类药物、生物活性研究等领域[6],如在临床上主要用于治疗帕金森综合症[7],促进转氨酶进行转氨作用提高体内多巴胺的含量[8]。
磷酸吡哆醛一般以盐酸吡哆醇(Vitamin B6,VB6)为原料合成。王淑美[9]用二氧化锰将VB6氧化后与二氯化镍反应制得吡哆醛螯合物,该螯合物磷酸化后通过阳离子交换树脂生成磷酸吡哆醛。该方法反应时间长,二氧化锰大大过量,分离方法不利于工业化生产[10]。黄金龙[11]等先用二氧化锰氧化VB6得到盐酸吡哆醛,后者与胺反应生成吡哆醛席夫碱。将席夫碱用多聚磷酸酯化后水解得到磷酸吡哆醛。该方法在氧化VB6过程中,需用浓硫酸调节反应体系的pH值,操作繁琐;吡哆醛席夫碱用多聚磷酸酯化,搅拌困难;席夫碱磷酸酯水解后,磷酸吡哆醛很难从水溶液中结晶。曹一丁等[6]利用一种化学与生物联合催化的方法将盐酸吡哆醇转化为磷酸吡哆醛,该方法环保,成本低廉,但总收率仅为20%左右。陈明等[12]通过二氧化锰氧化VB6生成醛,经席夫碱合成和磷酸酯化后水解得到磷酸吡哆醛,工业化前景值得探讨。
本研究以盐酸吡哆醇为原料,经图1所示路线合成磷酸吡哆醛,旨在通过对二氧化锰氧化盐酸吡哆醇的反应条件、盐酸吡哆醛席夫碱磷酸酯化条件,以及磷酸吡哆醛的结晶方法进行研究,探讨适合工业化生产的磷酸吡哆醛合成方法。
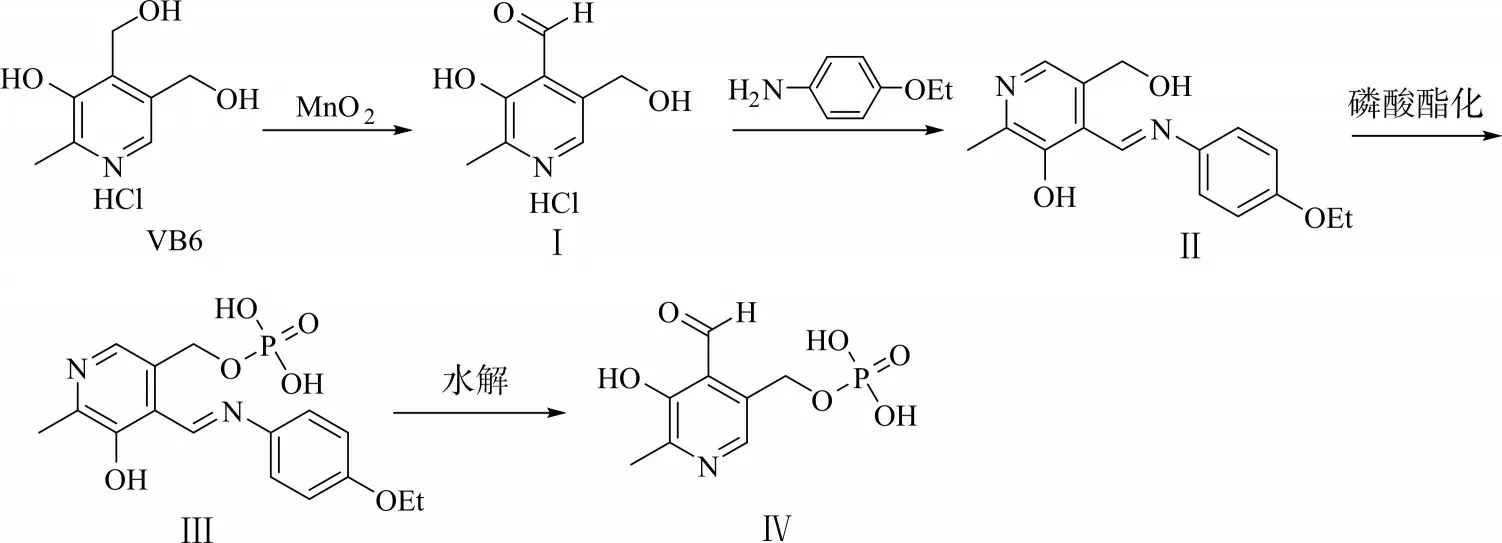
图1磷酸吡哆醛的合成路线Fig.1 Synthetic route of pyridoxal phosphate
1 实验部分
1.1 试剂与仪器
VB6(湖北惠生药业股份有限公司);浓盐酸,五氧化二磷,九水硫化钠(天津市大茂化学试剂厂);对乙氧基苯胺(上海阿拉丁生化科技股份有限公司);二氧化锰,多聚磷酸,氢氧化钠,磷酸(质量分数85%),乙酸乙酯,强酸性阳离子交换树脂(分析纯,国药集团化学试剂有限公司)。
RY-1熔点仪检测仪(天津天光光学仪器有限公司);DPX 300核磁共振仪(德国Bruker公司);EL III型元素分析仪(德国Vario公司);LC-20AT型高效液相色谱仪(HPLC)(日本岛津公司);Tracems 2000色谱-质谱联用仪(美国Finnigan公司);红外光谱仪(FTIR-650,天津)。
1.2 HPLC检测方法
每次均取0.1 mL反应液,用纯化水稀释到1.0 mL。再用一个干净的注射器连接一个0.22μm的筒式过滤器过滤样品。液相条件:C18(5μm,250 mm×4.6 mm);流动相:V(磷酸缓冲液)∶V(甲醇)=95∶5;缓冲溶液:20 mmol/L Na2HPO4·12H2O(用质量分数10%的磷酸调剂pH为7.0);流速:1.0 mL/min;柱温:30℃;进样量:3μL;检测波长:254 nm。梯度洗脱。
1.3 实验步骤
1.3.1 盐酸吡哆醛(I)的合成在10 L三口烧瓶中加入500.0 g(2.432 mol)VB6和5.0 L的质量分数为6%盐酸溶液,搅拌至溶解,控制反应温度在15℃,加入211.6 g(2.432 mol)二氧化锰,0.5 h后,再加入211.6 g(2.432 mol)二氧化锰。继续反应6 h后,经HPLC检测,盐酸吡哆醇已完全转化,盐酸吡哆醛I的选择性为96.2%。抽滤,水洗。往滤液中加入612.9 g(2.554 mo1)九水硫化钠,搅拌0.5 h。抽滤,滤液直接进行下步反应。
1.3.2 盐酸吡哆醛席夫碱(II)的合成在10 L三口烧瓶中,搅拌下将400.3 g(2.918 mol)对乙氧基苯胺缓慢滴加到上一步制备的盐酸吡哆醛溶液中,控制温度在15℃。滴加完毕后继续搅拌1 h,有大量橙黄色固体生成。抽滤,固体依次用水和乙酸乙酯洗涤,干燥得到化合物II 560.4 g,以VB6计收率为80.5%。m.p.:201~202.5℃,与文献值一致[13]。1H NMR(400 MHz,DMSO-d6)δ:14.20(s,1H,pyridine-OH),9.23(s,1H,-CH=N-),8.06(s,1H,pyridine-H),7.58(d,2H,benzene-H),7.13(d,2H,benzene-H),5.49(s,1H,-OH),4.85(s,2H,-CH2OH),4.16(m,2H,-OCH2-),2.51(s,3H,pyridine-CH3),1.44(s,3H,-CH3)。
1.3.3 磷酸吡哆醛席夫碱(III)的合成在装有搅拌器和温度计的10 L三口反应瓶中,加入516.3 g(1.803 mol)化合物II、1 827.7 g(5.409 mol)多聚磷酸和54.8 g水,50℃反应8 h后,加入4 130.4 g冰水,在30℃下搅拌0.5 h。加入430.3 g(11.789 mol)浓盐酸和1.5 g活性炭,升温达到75℃反应10 min,降至室温。抽滤,滤液用质量分数35%的氢氧化钠溶液中和,析出大量的橙红色固体。抽滤,固体用水洗涤至滤液为浅黄色为止,不需干燥可直接用于下一步反应。干燥得化合物III,m.p.:205~207℃。1H NMR(400 MHz,DMSO-d6)δ:14.36(s,1H,pyridine-OH),9.22(s,1H,-CH=N-),8.05(s,1H,pyridine-H),7.57(d,2H,benzene-H),7.03(d,2H,benzene-H),5.17(s,2H,-CH2),4.08(m,2H,-OCH2-),2.45(s,3H,pyridine-CH3),1.35(s,3H,-CH3)。EA(C16H19N2O6P),Calcd:C 52.46,H 5.23,N 7.65;Found:C 52.14,H 5.19,N 7.30。
1.3.4 磷酸吡哆醛(IV)的合成将上一步所得橙红色固体加入到5 L三口烧瓶瓶中,加2 706.0 mL 2 mol/L的氢氧化钠溶液,室温下搅拌2.0 h。反应液用乙酸乙酯萃取2次(1 500 mL×2)后,往水相中加入强酸性阳离子交换树脂1 500.0 g,搅拌1.0 h。抽滤,滤液冷冻干燥,粗产品经丙酮/乙醇(2∶1,V/V)打浆后得到淡黄色固体256.2 g,纯度为99.5%(HPLC检测),以化合物III计收率为76.6%。m.p.:141~142℃;MS(m/z):248.12[M+H+];IR(KBr)υ:3 368,2 927,1 632 cm-1.1H NMR(400 MHz,DMSO-d6)δ:10.36(s,1H,pyridine-CHO),8.09(s,1H,pyridine-H),5.17(s,2H,-CH2-),2.41(s,3H)。
2 结果与讨论
2.1 化合物I的合成
化合物I是合成磷酸吡哆醛的重要中间体,由VB6氧化制得。何地平等[13]将超声波辐射用于活性二氧化锰氧化吡哆醇的反应中;施湘君等[14]采用tempo氧化盐酸吡哆醇。钌配合物[15]以及钨酸铋[16-18]分别作为催化剂,用于双氧水氧化盐酸吡哆醇反应中。但这些氧化反应要么氧化体系复杂,不易操作,要么反应成本高,不适合工业化。本研究中采用二氧化锰为氧化剂,考察了盐酸吡哆醇与二氧化锰的摩尔比、盐酸溶液浓度、反应时间和温度(如表1所示)对氧化反应的影响,通过四因子三水平正交试验实验对氧化反应条件进行了优化,结果如表2所示。
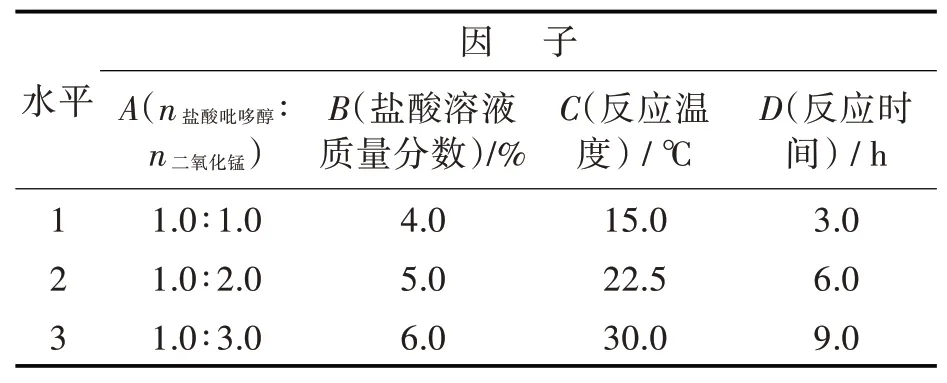
表1 MnO2氧化VB6正交试验方案Tab.1 Orthogonal testing program for oxidation of VB6 by MnO2
Tab.2 Orthogonal test results of oxidation of VB6 by MnO2
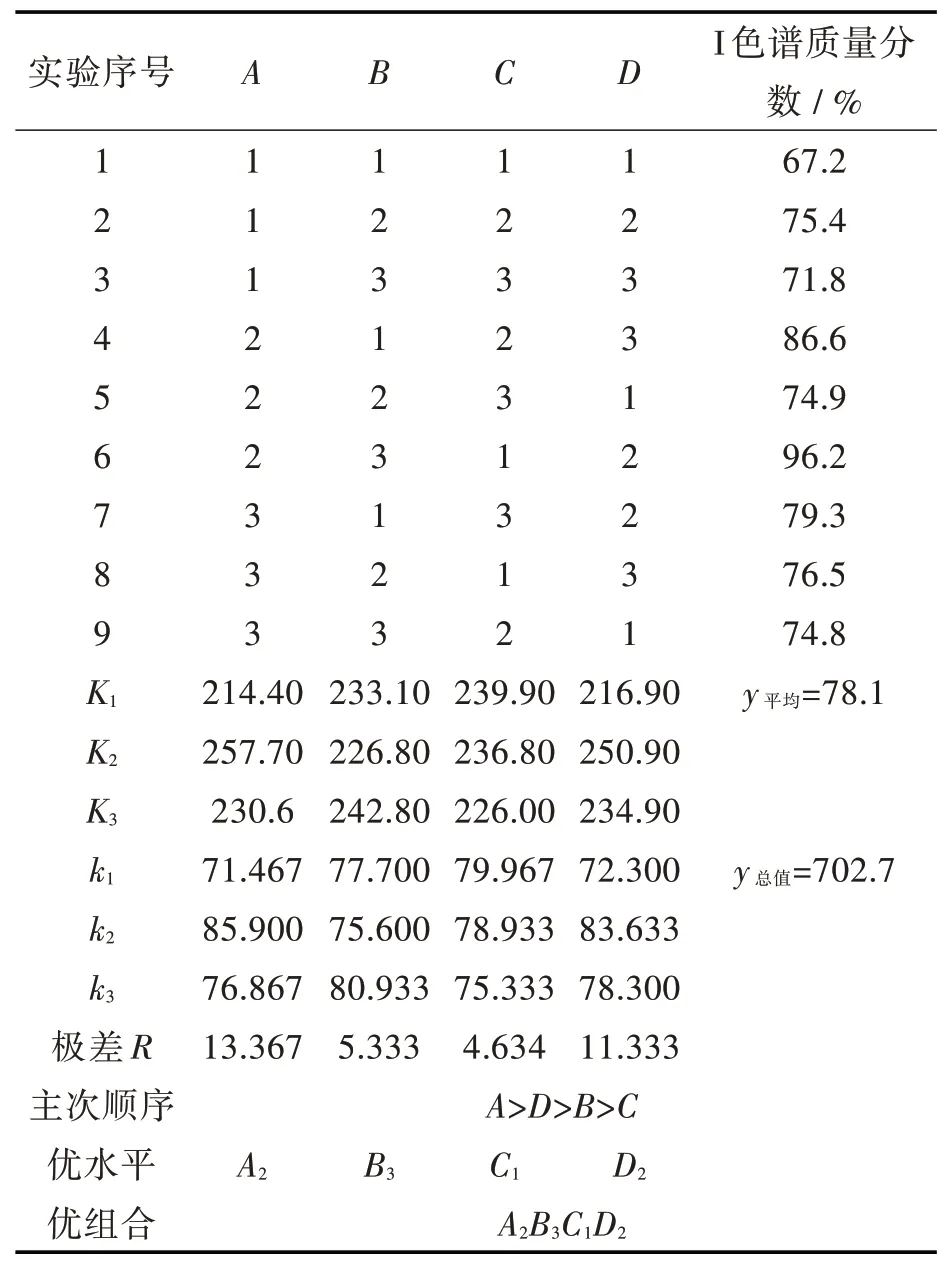
表2 MnO2氧化VB6正交试验结果
从计算出的K值得出A2、B3、C1、D2最佳。通过公式计算出本实验中各Rj的值得到:R1>R4>R2>R3,表明各因子A、D、B、C对氧化实验的影响逐渐减小。因此盐酸吡哆醇氧化反应的最优工艺条件为:A2B3C1D2,即盐酸吡哆醇与二氧化锰的摩尔比为1.0∶2.0,反应在质量分数6% HCl溶液中15℃下进行6 h。
二氧化锰氧化盐酸吡哆醇是一个放热反应,需在低温下进行。同时,酸性条件有利于VB6的选择性氧化,因为VB6吡啶环上氮原子质子化后,吡啶环电子云密度降低,不利于5-羟甲基的氧化。二氧化锰的用量也影响盐酸吡哆醛的生成。当n盐酸吡哆醇∶n二氧化锰=1∶1时,溶液中盐酸吡哆醛的最高含量为75.4%;当n盐酸吡哆醇∶n二氧化锰=1∶2时,盐酸吡哆醛的最高含量为96.2%;当n盐酸吡哆醇∶n二氧化锰=1∶3时,盐酸吡哆醛的最高含量为79.3%。由此可见MnO2过量过多,盐酸吡哆醛会过氧化。当反应进行6 h后,反应溶液中盐酸吡哆醛的含量不断降低,表明氧化时间过长,盐酸吡哆醛也会过氧化。
与文献[9]相比,本研究中二氧化锰的用量减少了1倍,反应温和,氧化反应时间由36 h缩短至6 h,收率由72.4%提高到96.2%。同时,也避免了文献[10]中反应溶液需用硫酸调节pH值的繁琐过程。氧化后的滤液中含有重金属离子Mn2+,实验通过加入Na2S生成MnS沉淀除去。
2.2 化合物II的合成
吡哆醛分子中吡啶环上4-甲酰基易被氧化,需在5-羟甲基磷酸酯化前将甲酰基保护起来。将醛转化为席夫碱是保护羰基的常用方法。芳香胺与吡哆醛反应生成席夫碱固体,由于具有芳香共轭体系,该席夫碱在酸性水溶液中稳定,不易水解。为提高苯胺氨基的亲核性,可以在苯环氨基的对位上引入给电子基团,如乙氧基、甲氧基、烷基等[19]。但若反应溶液的酸性过强,芳胺的—NH2基团会被质子化,亲核性减弱,降低席夫碱收率。实验中,将硫化钠处理后的滤液控制pH=5.0~6.0,加入对乙氧基苯胺与I反应,生成橙黄色固体,易于分离,以原料VB6计,吡哆醛席夫碱的收率达80%以上。
2.3 化合物III的合成
磷酸酯主要由醇与磷酸化试剂发生反应制得,常用的磷酸化试剂有H3PO4、P2O5、POCl3、PCl3、多聚磷酸等[20-21]。磷酸是三元酸,酯化产物是单烷基酯、双烷基酯和三烷基酯的混合物。以P2O5为磷酸化试剂,主要生成单烷基酯和双烷基酯。而PCl3和POCl3的腐蚀性较强,会腐蚀生产设备,不适于工业化生产。用多聚磷酸或P2O5/H3PO4为磷酸化试剂,产物主要为单烷基酯,能降低双烷基酯和三烷基酯的生成[22]。本研究以多聚磷酸和P2O5/85%H3PO4作为吡哆醛席夫碱的磷酸化试剂,对酯化反应体系进行改进,结果如表3所示。
从反应结果可以看出,化合物II与磷酸化试剂P2O5/质量分数85%H3PO4在50℃反应8 h,化合物III的收率最高达到72.8%,但P2O5易吸水,操作不便。多聚磷酸为粘稠状液体,与化合物II混合后形成粘稠物,搅拌很困难。为解决这一问题,林伟等[23]先将化合物II加入到含磷离子液体中,再与酸反应,收率达80%,含磷离子液体可循环使用。本实验向反应体系中加入DMF或DMSO溶剂,以及利用甲苯分水回流,虽然搅拌问题解决了,但多聚磷酸不溶于有机相,酯化反应收率低。在向反应体系中加入少量水后,多聚磷酸的流动性得以改进,反应混合物可以充分搅拌,且收率显著提高到85%以上。考虑到后处理,化合物II与多聚磷酸摩尔比为1∶3,反应在50℃进行8 h为化合物II磷酸酯化最优条件。
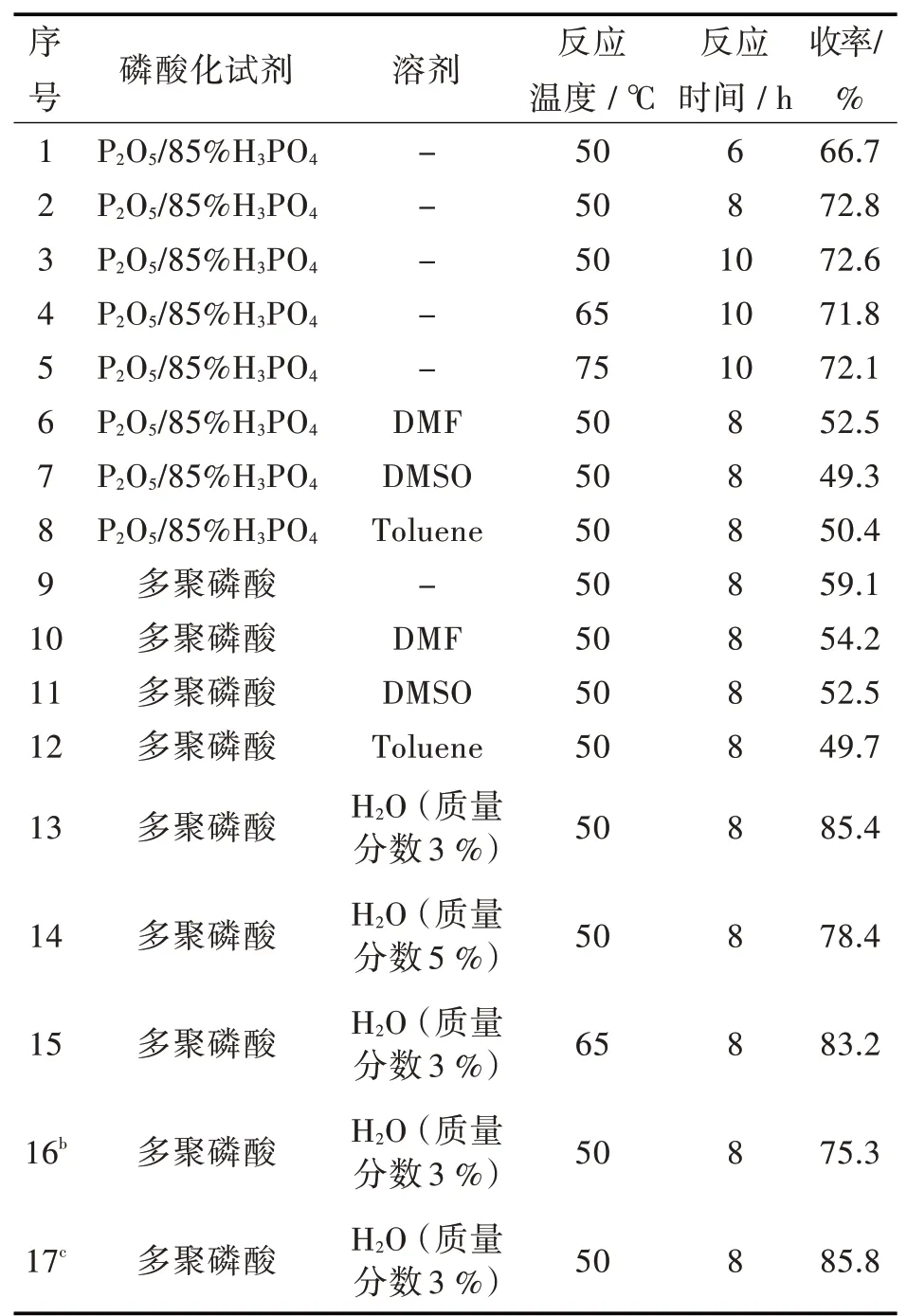
表3磷酸化试剂对吡哆醛席夫减磷酸酯化的影响aTab.3 Effect of phosphating reagents on phosphoric esterification of pyridoxal schiff basea
酯化反应完成后,在酸性条件下将多聚磷酸酯化产物加热水解。由于终产物磷酸吡哆醛在水溶液中温度高于40℃时极不稳定,而不加活性炭脱色又会影响产品质量,因此,本研究将脱色步骤与多聚磷酸酯化产物水解反应(75℃)同时进行。脱色后的滤液冷却后析出棕红色磷酸吡哆醛席夫碱固体。
2.4 化合物IV的合成
化合物III在碱性条件下水解得到磷酸吡哆醛的钠盐和对乙氧基苯胺,后者用乙酸乙酯萃取分离后可循环使用。磷酸吡哆醛分子中含有两性基团,在酸性或碱性条件下都能生成水溶性物质,因此,pH值是影响磷酸吡哆醛能否从水溶液中析出的重要因素。实验发现,用盐酸调节水溶液的pH至1.0到5.0之间,保持温度在0℃下均未有晶体析出。加入少许晶种诱导析晶,仅有少量淡黄色晶体析出,收率低于10%。在40℃以下将溶液减压浓缩至1/3体积后,析出深褐色固体,但其中化合物IV含量很少。可以看出,加热会使磷酸吡哆醛氧化。向水溶液中加入甲醇,仅析出NaCl固体。
鉴于以上实验结果,本研究先使用强酸性阳离子交换树脂将水溶液调酸,再将滤液冷冻干燥,得到淡黄色粉状磷酸吡哆醛产品,收率达90.1%。采用冷冻结晶方法,避免了磷酸吡哆醛被氧化,而利用离子交换树脂代替盐酸调节pH值,溶液中不会有无机盐存在,提高了冻干产品的质量。阳离子交换树脂经活化后可重复使用。
3 结论
本研究通过正交实验优化了二氧化锰氧化VB6的工艺条件,并利用MnS沉淀法除掉了溶液中的重金属Mn2+。利用含水(质量分数3%)的多聚磷酸对吡哆醛席夫碱进行酯化,改善了搅拌条件并显著提高了磷酸吡哆醛席夫碱的收率。采用阳离子交换树脂酸化磷酸吡哆醛席夫碱的水解溶液,并将溶液冷冻干燥制备磷酸吡哆醛,解决了磷酸吡哆醛从水溶液中结晶困难和含盐高的问题。整个合成路线反应条件温和、操作简便,磷酸吡哆醛的总收率达到61.6%,易于工业化生产。
