铁死亡调控机制及其在肿瘤治疗中的研究进展
2020-08-12阿依加马力麦麦提梁靓管晓翔
阿依加马力·麦麦提,梁靓,管晓翔,3#
1南京大学医学院,南京 210093
2江苏省人民医院句容分院血液肿瘤科,江苏 句容 212400
3江苏省人民医院肿瘤科,南京 210029
2012年,Dixon等[1]首次发现铁死亡。铁死亡在形态、生化和遗传上与其他为人熟知的自噬和凋亡不同。细胞凋亡中的染色质固缩和边缘化及聚ADP核糖聚合酶1[poly(ADP-ribose)polymerase 1,PARP1]裂解、线粒体释放的细胞色素c或caspase 3裂解、细胞自噬中的双层膜结构的自噬小泡、细胞器的肿胀、核膜和细胞膜的溶解等典型的特征在铁死亡中均没有出现。此外,由于发生铁死亡时,细胞膜或细胞器膜脂质过氧化和氧化应激,导致质膜选择渗透性丧失,线粒体形态发生特殊而独特的变化,包括线粒体嵴减少或消失、线粒体外膜破裂、线粒体膜浓缩[2]。
研究表明,人和动物不同的生理和病理应激状态均可引起铁死亡[1]。铁死亡可消除恶性细胞的适应性特征,去除无法获取关键营养因子或受感染及因环境改变而损伤的细胞,在抑制肿瘤发生中起关键作用。经典的氧化应激途径是诱发铁死亡的一个重要原因。虽然肿瘤细胞处于持续的氧化应激状态,但铁死亡潜在的分子机制仍未清楚。本文对肿瘤细胞中铁死亡的发生和调控机制进行综述,并详细介绍以铁死亡为基础的肿瘤治疗所面临的机遇和挑战,为肿瘤治疗探索具有临床价值的新策略。
1 铁死亡调控机制
近10年来,活性氧自由基(reactive oxygen species,ROS)一直是肿瘤研究领域的热点。细胞内的ROS主要由线粒体呼吸链产生,内质网、还原型烟酰胺腺嘌呤二核苷酸磷酸(reduced nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶复合体(NADPH oxidase,NOX)等也可以通过一系列化学反应产生ROS。正常情况下,氧化-抗氧化体系将ROS维持在一定的范围,超过正常范围则会导致肿瘤。铁死亡过程的主要特点是脂质活性氧(lipid-ROS,L-ROS)和来自铁代谢的致命ROS的积累,且可通过铁螯合剂和脂质过氧化抑制剂抑制。谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)、热休克蛋白β1(heat shock protein β1,HSBP1)和核因子相关因子 2(nuclear factor-f2,Nrf2)通过限制ROS产生和减少细胞对铁的摄取而对铁死亡起负调控作用。NADPH氧化酶和P53通过促进ROS产生发挥正调控作用[2](图1)。
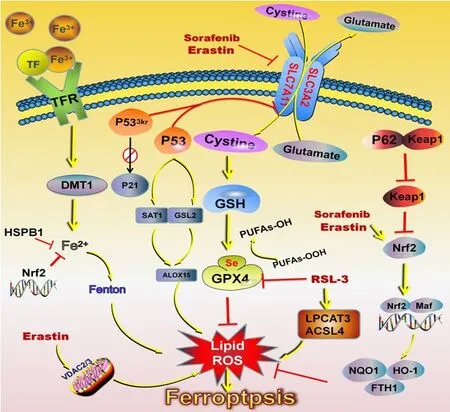
图1 铁死亡调控机制
1.1 铁死亡主要机制
L-ROS积累主要由GPX4活性丧失引起[3],两种不同的机制介导此过程:①是抑制胱氨酸/谷氨酸逆转运蛋白体(system xc-),通过system xc-细胞内的谷氨酸与细胞外的胱氨酸按照等比互换,可间接诱导GPX4失活。半胱氨酸(胱氨酸的还原形式)是谷胱甘肽(glutathione,GSH)合成的前体,GSH是GPX4发挥磷脂过氧化物酶活性并催化脂质过氧化物还原过程的主要辅助因子。因此,抑制system xc-会导致GSH耗竭并随后使GPX4失活(图1),最终导致L-ROS聚集从而引发铁死亡。②是铁死亡诱导剂直接抑制GPX4的酶促活性,在铁死亡的概念提出之前,研究者就发现了铁死亡诱导因子。2003年,研究发现了突变型大鼠肉瘤癌基因(rat sarcoma oncogene,RAS)表达的人包皮成纤维细胞BJeLR中参与合成致死的诱导因子Erastin[4]。system xc-是由溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)和溶质载体家族 3成员 2(solute carrier family 3 member 2,SLC3A2)组成的,研究证明,Erastin通过抑制SLC7A11而间接抑制GPX4的活化,破坏了细胞内氧化还原的稳定性及L-ROS聚集。随后,在2008年的另一项高通量小分子筛选研究中鉴定了RSL3,可以直接抑制GPX4的活性以非凋亡的形式选择性杀死BJeLR细胞[5](图1),且小分子抑制剂不能逆转RSL诱导的细胞死亡。相反,抗氧化剂和铁螯合剂可阻断RSL诱导的细胞死亡。因此,也进一步证明铁死亡是一种铁依赖性细胞死亡。
1.2 铁代谢
已经证实,与铁代谢相关的基因是铁死亡过程中的关键介质(图1),如转铁蛋白(transferrin,Tf)、转铁蛋白受体1(transferrin receptor 1,TfR1)、铁卟啉(iron porphyrin,FPN)、二价金属离子转运体1(divalent metal transporterq, DMT1, 又 称SLC11A2)、铁蛋白重链(ferritin heavy chain 1,FTH1)、铁蛋白轻链(ferritin light chain,FTL)等。Fe3+通过TfR1进入细胞,定位于内胚层,并通过金属还原酶家族3(six-transmembrane epithelial antigen of the prostate 3,STEAP3)的铁还原酶还原为Fe2+。最后,SLC11A2促进Fe2+释放到细胞质上的不稳定铁池中,通过Fenton反应介导ROS产生[6],多余的铁储存在铁蛋白中。膜铁转运蛋白(铁外排泵,也被称为SLC11A3)将Fe2+氧化为Fe3+输出。
学者对发生铁依赖性死亡的细胞研究发现,TfR1增加和铁蛋白(FTL和FTH1)表达下调,是铁死亡的细胞中铁超载的原因。铁螯合剂通过限制铁超载从而抑制铁死亡,而供应外源性铁可促进铁死亡。研究显示,蛋白62(protein 62,p62)/Kelch样ECH联合蛋白1(kelch-like ECH-associated protein 1,Keap1)/Nrf2抗氧化信号通路是通过转录激活活性氧和铁代谢相关基因而抑制铁吸收[7](图1)。相反,HSP27的磷酸化阻断铁吸收从而抑制铁死亡。蛋白激酶 C(protein kinase C,PKC)介导HSPB1的磷酸化,并通过抑制ROS产生和减少铁吸收从而成为铁死亡的负调节剂[8]。
1.3 P53参与铁死亡
研究表明,P53与铁死亡机制相关(图1),P53可直接抑制system xc-的关键成分SLC7A11的转录而抑制细胞对胱氨酸的摄取。此外,乙酰化缺陷突变体小鼠缺乏细胞周期阻滞、凋亡或衰老的功能,但可以通过抑制SLC711A的表达而诱导铁死亡,仍具有肿瘤抑制功能[9]。在某些特定条件下,P53的表达可能促进、限制或延缓铁死亡的开始。P53在细胞铁依赖性死亡过程中的这些作用是通过不同机制实现的,如P53对代谢基因转录、翻译后调控或对P53-P21轴的影响等[10]。P53以细胞特异性或环境依赖性的方式对铁死亡的双向调节需要进一步研究。
1.4 其他机制
Erastin除通过抑制system xc-和铁通过Fenton反应介导ROS产生外,还有很多种途径产生ROS并引发铁死亡。电压依赖性阴离子通道2(voltagedependent anion channel 2,VDAC2)/VDAC3信号转导通路在线粒体外膜上控制腺苷二磷酸(adenosine diphosphate,ADP)、磷脂酰肌醇(phosphatidylinositol,PI)和腺苷三磷酸(adenosine triphosphate,ATP)的跨膜流动。微管蛋白可以通过阻断VDAC2/VDAC3信号转导通路抑制线粒体代谢,从而促进有氧糖酵解(是肿瘤细胞主要的能量来源)。Erastin具有直接抑制微管蛋白的功能,使VDAC2/VDAC3处于开放状态并恢复线粒体代谢,诱导活性氧的产生[11](图1),最终诱导细胞以独特的铁依赖性、非凋亡形式死亡。此外,铁死亡诱导剂可以诱导线粒体上的戊糖磷酸途径(pentose phosphate pathway,PPP)产生 NADPH,通过 NOX氧化酶将电子转移并氧转化为过氧化物。此外,谷氨酰胺转化为α-酮戊二酸(α-ketoglutaric acid,α-KG)可产生ROS。
德国学者研究发现,脂代谢相关的长链脂酰辅酶A合成酶家族成员4(long-chain acyl-CoA synthetase 4,ACSL4)是铁死亡中的关键蛋白之一[12]。花生四烯酸和肾上腺酸是多不饱和脂肪酸底物,ACSL4可酰基化花生四烯酸和肾上腺酸,使其成为膜磷脂的一部分,易被氧化成脂质ROS,而GSH和GPX4对其有还原作用。RSL3可以直接抑制GPX4,导致这些脂质ROS的积聚和脂质过氧化,从而引发铁死亡[12](图1)。半胱氨酰转运RNA(transfer RNA,tRNA)合成酶可以剥夺胱氨酸,减少GSH的合成,使GPX4失活,从而产生ROS诱导铁死亡[13]。
1.5 非编码RNA 在细胞铁死亡中的作用
越来越多的学者认为,微小RNA(microR NA,miRNA)和长链非编码 RNA(long non-coding RNA,lncRNA)是调控铁死亡的关键介质(表1)。研究发现,肺腺癌中的P53RRA异常低表达。P53与RNA结合蛋白(G3BP1)结合[14],P53RRA与G3BP1的RNA识别基序结构域相互作用,将P53与其分离并使细胞核中的P53保存更多,通过P53途径增加铁和脂质ROS的浓度。从而得知,P53RRA-G3BP1是肺腺癌中诱导铁死亡的一种新的机制,肺腺癌组织中P53RRA表达水平越低,肺腺癌患者的预后就越差。
在黑色素瘤细胞系G-361和A375中,miRNA-9可以通过靶向谷氨酸-草酰乙酸转氨酶1(glutamate oxaloace-tate transaminase 1,GOT1)来抑制铁死亡。GOT1是分解葡萄糖,并将其转化为α-KG的酶,其可促进ROS的积累并诱导铁死亡。另一项研究表明,miRNA-137可直接抑制SLC1A5来阻止铁死亡,而SLC1A5是葡萄糖摄取的主要受体。因此,通过敲除miRNA-9和miRNA-137,可提高α-KG的水平并促进葡萄糖的摄取,从而提高肿瘤细胞对铁死亡的敏感性。
SLC7A11在铁死亡的负调控中发挥关键作用。研究表明,miRNA-375、miRNA-27a、miRNA-26b和As-SLC7A11(反义lncRNA)能抑制SLC7A11的转录,降低其蛋白强度[15]。因此,这些miRNA可以通过靶向SLC7A11来促进铁死亡。
由于Nrf2具有抑制铁吸收、限制ROS生成和上调SLC7A11表达的功能,因此,Nrf2也是铁死亡的重要抑制剂。首先,miRNA-7和miRNA-200a通过抑制Keap1的表达,激活Nrf2途径[16];相反,miRNA-28以不依赖Keap1的方式抑制Nrf2的表达[17]。其次,miRNA-101和miRNA-455均可通过靶向Cullin-3(Cul3)来促进Nrf2的表达[18]。最后,miRNA-153、miRNA-142-5p、miRNA-27a、miRNA-144、miRNA-93、miRNA-34a、miRNA-365-1、miRNA-193b和 miRNA-29-b1[18]可通过不同机制降低Nrf2的水平。这些结果表明,miRNA可以通过调节Nrf2的表达来影响铁死亡。
根据现有的研究结果得知,miRNA还参与铁的转运、储存、利用和吸收的调控。miRNA-20a和miRNA-485-3p可以通过靶向FPN基因来降低铁产量[19],miRNA-210和miRNA-152抑制TfR1的表达,从而减少对Tf的吸收。同时,miRNA-200b和miRNA-Let-7d的表达分别通过抑制FTH和DMT1-IRE的表达有效减少了铁的积累。
2 铁死亡是程序性细胞死亡的扩展网络
近5年,我们对铁死亡的认识得到了惊人的提高,铁死亡与细胞凋亡、自噬和其他程序性坏死之间的作用都已进行了初步研究。

表1 与铁死亡相关非编码RNA的表达情况
2.1 铁死亡和细胞凋亡:转换、协同还是拮抗作用?
越来越多的研究表明,P53除通过细胞周期停滞和诱导细胞凋亡来预防肿瘤发生外,还可以在某些条件下诱导铁死亡。张先正教授等设计了一种新型的P53复合物——MON-P53[20]。MON引起Fenton反应调节ROS积累,P53抑制SLC7A11诱导GSH清除引起铁死亡,还能调节凋亡途径引起细胞凋亡,从而限制肿瘤的生长及延长肿瘤切除小鼠的寿命。因此,该方法将指导铁死亡和细胞凋亡混合抗肿瘤治疗。
青蒿琥酯和Erastin之类的铁死亡诱导因子可以诱导未折叠的蛋白应答,该蛋白通过C/EBP同源蛋白(CAAT/enhancer binding protein homologous protein,CHOP)促进P53上调促凋亡因子(P53 upregulated modulator of apoptosis,PUMA)的表达[21]。PUMA可以增强肿瘤坏死因子相关凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)的表达来促进细胞凋亡。通过观察TRAIL诱导的细胞凋亡作用可以证实铁死亡诱导剂与TRAIL联合能明显提高对肿瘤细胞的杀伤作用。
另一种观点认为,刺激铁死亡相关的代谢在生化方面会抑制凋亡的发生。由于胱氨酸减少而发生铁死亡的细胞,其细胞内GSH含量约为正常水平的10%。GSH的还原能力是caspases 3和caspases 8激活的必要条件,因此,细胞缺乏GSH不能正确激活caspase,从而抑制细胞凋亡。
2.2 自噬降解铁蛋白可以促进铁死亡
自噬是一种依赖溶酶体的降解途径。自噬途径的激活可以降解铁蛋白,然后触发肿瘤细胞的铁死亡[22],也可以通过溶酶体ROS的产生促进铁死亡,还可以通过NCOA4介导的铁蛋白自噬为铁死亡提供不稳定铁。敲除自噬相关基因Atg5和Atg7通过减少脂质过氧化和细胞内Fe2+水平,从而限制铁死亡。
最近的研究表明,细胞因子信号转导抑制因子1(suppressor of cytokine signaling 1,SOCS1)是P53基因的直接调节和调控细胞衰老所必需的重要因子,而P53可抑制SLC7A11转录,从而抑制GSH的摄取。SOCS1可以通过调控P53,使细胞对铁死亡敏感[23]。尽管不同类型调控细胞死亡的机制具有不同的形态学和生化特征,但在调控因子与这些不同过程的组成部分之间仍存在一些干扰。
3 铁死亡在不同肿瘤治疗中的调控机制
NCI-60是一组由8种不同组织类型组成的不同肿瘤细胞系,其中弥漫性大B细胞淋巴瘤和肾细胞癌细胞比其他6个组织(乳腺癌、肺癌、结肠癌、黑素细胞癌、中枢神经系统癌和卵巢癌)的肿瘤细胞更易发生Erastin诱导的铁死亡。由于基本代谢状态的不同,不同细胞系对铁死亡的敏感性也不同。大量研究证实,如阿糖胞苷、顺铂、阿霉素和替莫唑胺等化疗药物与Erastin联合应用对其抗肿瘤活性有明显的协同作用,患者的预后优于单纯传统化疗。
3.1 肝细胞癌
铁死亡是索拉非尼治疗肝细胞癌的潜在机制之一。肝细胞癌组织中视网膜母细胞瘤(retinoblastoma,Rb)水平明显降低,索拉非尼处理的肝细胞癌细胞中Rb蛋白更容易失活。抑制system xc-和GSH耗竭是索拉非尼诱导铁死亡的主要机制。Rb蛋白失活导致线粒体中ROS浓度增加并增强肝细胞癌细胞的氧化应激反应,提高其对铁死亡的敏感性。MT-1G是肝细胞癌中一种新型的铁死亡负调节剂,敲除MT-1G可通过增加脂质过氧化作用和GSH耗竭而导致铁死亡。低密度脂蛋白-二十二碳六烯酸纳米颗粒通过铁死亡途径导致肝细胞癌细胞死亡。过度活跃的P62/keap1/Nrf2途径由于Nrf2的靶基因,包括血红素加氧酶-1(HO-1)、FTH1和醌氧化还原酶1[NAD(P)H:quinine oxidoreductase 1,NQO1]可以直接抑制ROS的积聚(图1),从而削弱铁死亡过程[24]。
3.2 胃癌
Hao等[25]发现,Erastin可以诱导胃癌细胞发生铁死亡。铁死亡抑制剂(F-1和L-1)可以抑制CDO1,从而恢复GPX4的表达和GSH水平,最终减少ROS的生成来逆转Erastin诱导的铁死亡。CD44是在肿瘤干细胞中表达的黏附分子。研究发现,CD44突变体(CD44v)可与system xc-相互作用,调控细胞内GSH的水平。因此,CD44高表达的人胃肠道肿瘤细胞GSH合成能力高,进而可以阻断ROS诱导的应激信号传导,使胃癌细胞对铁死亡产生抵抗[26]。
3.3 卵巢癌
卵巢癌细胞对铁死亡的敏感性很高,因为其肿瘤起始细胞TICs中铁水平过高,TFR1过表达,铁外排泵FPN水平过低[27]。青蒿琥酯是一种耐受性良好的抗疟疾药物,也具有抗肿瘤活性。研究发现,采用青蒿琥酯处理的卵巢癌细胞发生ROS与铁依赖性细胞死亡,并且铁死亡抑制剂F-1可有效阻止这一过程[28]。
3.4 乳腺癌
Ma等[29]研究显示,西拉美新和拉帕替尼通过增加铁依赖性ROS的产生来诱导铁死亡。CDO1过表达会降低乳腺癌细胞中GSH水平,导致ROS的进一步积累从而加剧铁死亡[30]。相反,MUC1-C可以通过与CD44v形成复合物来增加半胱氨酸的摄取并上调GSH的表达,可抑制乳腺癌细胞铁死亡过程。三阴性乳腺癌中ACSL4的表达水平较高,其可以通过参与脂质ROS的积聚和脂质过氧化而引发铁死亡。
3.5 肺癌
Erastin在KRAS突变的A549细胞中首次诱导肺癌细胞铁死亡。研究显示,Erastin通过减少GSH和GPX4灭活引发铁死亡。顺铂也是一种铁死亡诱导剂,其机制与Erastin相似。此外,顺铂联合Erastin治疗肺癌的抗肿瘤活性存在明显的协同作用[27]。半胱氨酸脱硫酶(cysteine desulfurase,NFS1)作为铁硫簇生物合成酶,通过维持铁硫辅因子,可以在高氧张力下保护细胞免受铁死亡。
3.6 横纹肌肉瘤
横纹肌肉瘤(rhabdomyosarcoma,RMS)是一种影响儿童和青少年的肌源性肿瘤,对其发病机制的研究甚少。固有的RAS/ERK信号通路是发生RMS的主要驱动因素之一,此通路的激活与肿瘤侵袭性和氧化应激水平相关。最近的研究表明,高表达的GSH在RMS细胞生长和RMS多药耐药性研究中至关重要。铁死亡诱导剂RSL3和Erastin可使GPX4耗竭并降低GSH水平,从而诱导RMS13细胞铁死亡并可以明显阻止肿瘤的进展。
3.7 血液恶性肿瘤
一项研究从各种组织收集8种细胞系,其中弥漫性大B细胞淋巴瘤细胞是对铁死亡诱导剂最敏感的细胞之一[3]。研究证明,GPX4过表达是弥漫性大B细胞淋巴瘤预后的独立预测因子。此外,体外分析表明,GPX4过表达细胞对ROS诱导的细胞死亡具有抵抗力。相反,GPX4基因敲除细胞对ROS诱导的细胞死亡敏感。上述研究表明GPX4调节ROS诱导的细胞死亡过程[31]。
急性髓系白血病(acute myeloblastic leukemia,AML)是成年人最常见的白血病类型。研究显示,Erastin以一种与RAS无关的方式增强AML细胞对化疗药物的敏感性。Erastin剂量依赖性地诱导HL-60细胞(AML,NRAS_Q61L)中与铁死亡、凋亡、坏死和自噬相关的混合型细胞死亡。铁死亡抑制剂或细胞坏死抑制剂可防止Erastin诱导的细胞死亡,但Z-VAD-FMK(一种通用的caspase抑制因子)或氯喹(一种有效的自噬抑制因子)则不会防止。重要的是,小剂量Erastin明显增强了两种一线化疗药物(阿糖胞苷/阿糖胞苷和阿霉素)对HL-60细胞的抗肿瘤活性。总之,诱导铁死亡和坏死有助于Erastin抑制AML细胞生长和克服耐药性[32]。
4 小结与展望
综上所述,目前,根除残留或耐药肿瘤细胞在肿瘤治疗的相关研究中至关重要,铁死亡作为一种新型细胞死亡形式为肿瘤治疗提供了一个新的治疗策略。有研究报道,获得间充质细胞状态(如上皮-间充质转化或肿瘤干细胞)与肿瘤细胞转移及扩散、耐药和化疗抵抗有密切关系[33]。高间充质状态的肿瘤细胞存活依赖于GPX4,并且高间充质状态是获得性和非获得性抗靶向治疗的重要机制。阻断GPX4将直接导致铁死亡,持续存在的肿瘤细胞通过肿瘤休眠状态摆脱常规治疗的细胞毒性,此状态也对GPX4通路有相同的选择依赖性。因此,铁死亡可能是逆转肿瘤细胞耐药性的可行策略。铁死亡比细胞凋亡更具免疫原性,铁死亡细胞通过DAMP释放炎性相关的损伤分子(如HMGB1)促进炎症的发生[34],还可通过传递趋化性信号在肿瘤部位招募和激活免疫细胞,这说明铁死亡诱导剂很有可能对抗肿瘤免疫治疗有促进作用。
虽然铁死亡在肿瘤治疗中效益明显,但如何从分子水平上解释铁死亡与其他细胞死亡之间的相互联系,铁死亡是否跟细胞坏死一样增强细胞对宿主的免疫原性,从而引起适应性免疫反应;在临床前或临床试验中,是否通过铁死亡诱导剂来有效杀死肿瘤细胞等一系列问题仍需要进一步研究。
