高效液相色谱-串联质谱法同时测定蜂胶、蜂胶原料保健食品中的氯霉素、甲砜霉素与氟甲砜霉素
2020-08-03廖夏云刘常凯余仁连苏小婷
杨 黎,刘 星*,廖夏云,刘常凯,余仁连,苏小婷
(1.广西-东盟食品检验检测中心,广西 南宁 530021;2.广西中医药大学 药学院,广西 南宁 530021)
我国农业农村部最新颁布的第250号公告中,氯霉素及其盐、酯被列入食品动物中禁用的药品及其他化合物清单[1]。蜂胶作为一种动物源性食品,氯霉素及其盐、酯应禁止检出。2002年,蜂胶被我国卫健委公布为可用于保健食品原料的一种中药,随着对蜂胶抗氧化、免疫调节、抗菌、抗肿瘤、胰岛素抵抗等多种生物活性[2-5]的深入研究,蜂胶被开发成各种剂型的保健食品。氯霉素(CAP)、甲砜霉素(TAP)和氟甲砜霉素(FF)同属于氯霉素类药物,甲砜霉素和氟甲砜霉素为氯霉素的衍生物,药理活性类似,均具有广谱抗菌能力,常被用于预防和治疗蜜蜂细菌性疾病[6]。如在蜂产品原料的收获期违规使用此类药物,将导致蜂胶及蜂胶原料保健食品中氯霉素类药物的残留[7]。
我国蜂胶原料保健食品消费规模巨大,对蜂胶的主要关注点多在真伪鉴别[8-11]、药理作用[12-15]及有效成分研究[16-22]等方面,对其安全性指标关注较少。我国发布有关蜂胶的标准,仅对蜂胶的质量指标做了有限规定,在兽药残留等安全性指标上的检验方法及规定几乎空白,因此有必要建立蜂胶及蜂胶原料保健食品中兽药残留的检测方法。国内已有关于蜂胶原料或蜂胶提取物中氯霉素残留测定的文献报道[23-25],肖利龙等[26]采用液液萃取结合高效液相色谱-串联质谱(HPLC-MS/MS)法同时测定蜂胶中的氯霉素、甲砜霉素和氟甲砜霉素;国外Boboni等[7]建立了意大利市售蜂胶提取物中氯霉素的液相色谱-串联质谱分析方法。但以蜂胶为原料的保健食品基质复杂,添加有油脂、成型辅料等成分,上述方法不一定适用于蜂胶原料保健食品。近年来,蜂胶原料保健食品中氯霉素等兽药残留已引起关注[27-28]。但目前国内外均无适用于蜂胶及蜂胶原料保健食品中氯霉素、甲砜霉素和氟甲砜霉素同时检测的方法,建立简单、有效的检测方法迫在眉睫。本文以0.1 mol/L盐酸为提取溶剂,经HLB与NH2固相萃取柱组合净化,采用HPLC-MS/MS同时测定蜂胶、蜂胶原料保健食品中氯霉素、甲砜霉素和氟甲砜霉素残留,可为蜂胶及蜂胶原料保健食品中兽药残留检测标准制订和蜂胶产品安全监管提供技术支撑。
1 实验部分
1.1 仪器、试剂与材料
ACQUITY UPLC I-Class高效液相色谱仪、Waters TQ-XS三重四极杆串联质谱仪(Waters公司);Mettler Toledo ML204电子分析天平(梅特勒-托利多仪器(上海)有限公司);多管式涡旋振荡器(德国Heidolph公司);Thermo Multifuge X3R低温冷冻高速离心机(美国Thermo公司);Organomation N-EVAP 112氮吹仪(美国Organomation公司);ZABN2000氮气发生器(南京日立产机有限公司)。
标准品:氯霉素(纯度99.9%)、甲砜霉素(纯度99.9%)、氟甲砜霉素(纯度99%),均购于德国Dr.Ehrenstorfer公司;乙腈、甲醇、正己烷、甲酸(色谱纯),均购于默克公司;25%氨水、盐酸、二氯甲烷(分析纯),均购于广东光华科技股份有限公司;Oasis HLB 固相萃取柱(6 mL/200 mg,Waters公司);Agela Cleanert NH2-SPE 柱(3 mL/500 mg,Agela公司)。
样品:蜂胶原料、蜂胶原料保健食品均为本地市售。
1.2 实验方法
1.2.1 标准溶液的配制分别准确称取适量氯霉素、甲砜霉素、氟甲砜霉素标准品,用乙腈溶解配制成100 μg/mL的标准储备溶液,-20 ℃冰箱保存。准确移取适量上述标准储备溶液,用乙腈配制成100 μg/L的混合标准溶液,-20 ℃冰箱保存。
取适量混合标准溶液,用0.1%甲酸水-乙腈(90∶10,体积比)配制成氯霉素、甲砜霉素、氟甲砜霉素质量浓度为0.10~20.0 μg/L的混合标准工作液,临用现配。
1.2.2 样品前处理蜂胶原胶、硬胶囊剂型、片剂:蜂胶原胶置于-20 ℃冰箱冷冻过夜,取出立即打碎成均匀粉末;硬胶囊剂型直接取胶囊内容物;片剂研磨成均匀的粉末作为样品。准确称取样品(2.00±0.05) g于50 mL离心管中,精密加入0.1 mol/L盐酸10.0 mL,置于涡旋混合器上高速涡旋2 min,超声5 min后以10 000 r/min离心10 min。将HLB固相萃取柱预先用5 mL甲醇和5 mL水活化,精密吸取5.0 mL上清液上样后,用5 mL水、5 mL 5%甲醇淋洗,负压抽干10 min。将NH2柱用5 mL甲醇活化,保持柱体湿润,串接在抽干后的HLB柱下,用5 mL甲醇、5 mL 4%氨化甲醇洗脱。收集洗脱液,50 ℃以下氮吹至干,用1.0 mL 0.1%甲酸水-乙腈(90∶10)复溶,过0.22 μm微孔滤膜,待测定。
软胶囊剂型:称取内容物(2.00±0.05) g于50 mL离心管中,精密加入0.1 mol/L 盐酸10.0 mL,加入10 mL正己烷,置于涡旋混合器上高速涡旋5 min,超声5 min后以10 000 r/min离心10 min,此时混合溶液分为3层,中间层为提取液,下层为固体,弃去上层正己烷层。将HLB固相萃取柱预先用5 mL甲醇和5 mL水活化,吸取中间澄清提取液5.0 mL上样后,用5 mL水、5 mL 5%甲醇淋洗,负压抽干10 min。后续步骤与其他3种样品相同。
1.3 分析条件
色谱柱:Waters ACQUITY UPLC®BEH C18柱(2.1 mm×100 mm,1.7 μm),柱温:40 ℃;进样量:1 μL;流速:0.4 mL/min;流动相:A为0.1%甲酸水,B为乙腈。梯度洗脱程序:0~3.0 min,10%~25%B;3.0~3.5 min,保持25%B;3.5~5.0 min,25%~75%B;5.0~5.5 min,保持75%B;5.5~6.0 min,75%~10%B;6.0~8.5 min,保持10%B。
离子源:电喷雾离子(ESI)源,负离子模式;扫描方式:多反应监测(MRM),分段采集;毛细管电压:-0.30 kV,离子源温度:150 ℃,脱溶剂温度:500 ℃,脱溶剂气流速:1 000 L/h,锥孔气流速:150 L/h,碰撞气(氩气)流速:0.15 mL/min,其他MRM参数见表1。
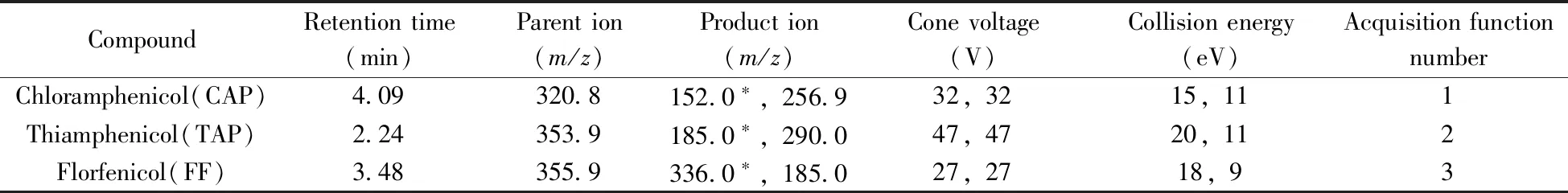
表1 目标化合物的质谱参数Table 1 MS parameters of chloramphenicol,thiamphenicol and florfenicol
2 结果与讨论
2.1 提取溶剂的选择
蜂胶原胶中含有大量的树脂、蜂蜡以及蜜蜂上腭腺分泌的粘液质等成分[29],软胶囊剂型添加了甘油、明胶、油脂等辅料,因此提取溶剂需将样品完全融化并减少共提取物。考察了乙酸乙酯、乙腈、0.5%氢氧化钙、0.1 mol/L EDTA-Mcllvaine缓冲液(pH 4.0)、4%醋酸铅、0.1 mol/L盐酸作为提取溶剂时对软胶囊剂型的效果。结果显示,采用乙酸乙酯和乙腈提取时,软胶囊剂型的提取液颜色很深,共提取干扰物多,回收率不理想;0.1 mol/L EDTA-Mcllvaine缓冲液(pH 4.0)不能使软胶囊剂型的内容物均匀分散,涡旋后内容物呈团块状,不利于目标成分溶出;0.5%氢氧化钙和4%醋酸铅能很好地溶解样品,但会破坏部分目标物,导致响应降低;0.1 mol/L盐酸能较好地溶解软胶囊剂型的内容物,且提取溶液颜色较浅,并可使目标化合物离子化,增强在HLB固相萃取柱上的吸附,实验最终采用0.1 mol/L盐酸为提取溶剂。另外,软胶囊剂型多以植物油脂(如大豆油等)作为辅料,提取时加入正己烷可除去样品中的脂溶性杂质,有助于提取和净化。
片剂和硬胶囊剂型主要添加固体类辅料,实验证明0.1 mol/L盐酸适用于片剂、硬胶囊剂型及蜂胶原料的提取。
2.2 净化方式的选择
固相萃取是兽药残留检测的常用净化手段,其中HLB柱填料为聚苯乙烯/二乙烯苯以及亲水性的吡咯烷酮基团,同时具有亲水性和疏水性,对各类极性和非极性化合物均具有吸附作用,能够去除样品中的不保留基质组分,如盐类、糖类、极性脂质和蛋白质等。实验发现,HLB柱上样后必须经水、5%甲醇淋洗步骤,否则试管底部会有大量未知粘稠物,影响复溶及目标物的回收率。比较了6 mL/200 mg和6 mL/500 mg两种规格HLB萃取柱对回收率的影响,结果显示两柱对目标化合物的回收率无明显影响,综合考虑成本,选择HLB(6 mL/200 mg)柱。NH2柱具有弱阴离子交换和正相保留作用,对脂肪酸、极性色素和糖具有很好的结合力,因此本实验在HLB柱后串接NH2柱,以去除基质中脂肪酸(如油酸、棕榈酸、亚油酸)、有机酸、部分色素、金属离子、糖类等干扰物,提高净化效果。
2.3 检测条件的优化
配制100 μg/L的混合标准溶液用针泵直接进样,分别对毛细管电压、离子源温度、脱溶剂温度、脱溶剂气流速、锥孔气流速、碰撞气(氩气)流速、锥孔电压、碰撞能量等质谱参数进行优化,以确定化合物的母离子和碎片离子,以响应强度较大的子离子作为定量离子,响应强度略小的子离子作为定性离子,典型的多反应监测谱图见图1。由不同基质加标的图谱可见,在本实验条件下目标化合物能与共流出物有效分离。

图1 空白软胶囊剂型基质添加目标分析物(10.0 μg/kg)的多反应监测(MRM)谱图Fig.1 MRM chromatograms of blank soft capsule matrix adding target substance of 10.0 μg/kg
2.4 基质效应评价
基质效应是由基质中的共提取干扰物与目标化合物竞争电离所致[30]。基质效应会影响液相色谱-串联质谱仪的灵敏度和精密度,从而影响定量准确性。本实验制备蜂胶原胶、片剂、硬胶囊剂型和软胶囊剂型4种不同基质的空白基质,分别配制质量浓度为0.10~20.0 μg/L的基质标准溶液以及相应质量浓度的溶剂标准溶液,外标法制作标准曲线。采用基质标准曲线的斜率/溶剂标准曲线的斜率评价基质效应[31],若比值在0.8~1.2范围内,认为基质效应不明显。结果表明,4种基质的基质效应为0.92~1.18,说明本方法具有良好的净化效果,基质效应不明显。
2.5 线性关系
按“1.2.1”配制质量浓度为0.10~20.0 μg/L 的混合标准工作液,以目标物的质量浓度(x,μg/L)为横坐标,相应峰面积(y)为纵坐标进行线性回归。结果显示,3种目标化合物在0.10~20.0 μg/L范围内线性关系良好,相关系数(r2)均大于0.999。
2.6 检出限与定量下限
在4种不同基质的阴性样品中添加0.5 μg/kg的目标化合物,采用本方法提取净化后进行测定,以信噪比S/N≥3计算方法的检出限,S/N≥10计算方法的定量下限。结果显示,蜂胶原胶、片剂和软胶囊剂型中3种目标物的检出限(LOD)为0.037~0.083 μg/kg,定量下限(LOQ)为0.12~0.28 μg/kg;硬胶囊剂型中的LOD为0.39~0.47 μg/kg,LOQ为1.28~1.57 μg/kg(见表2)。
2.7 回收率与相对标准偏差
分别对蜂胶原胶、软胶囊剂型、硬胶囊剂型和片剂样品进行加标回收实验,在4种不同的阴性基质中分别添加2.5、5.0、10.0 μg/kg 3个浓度水平的标准品,每个浓度5份,采用本方法进行处理和测定,计算3种目标化合物的回收率及相对标准偏差(RSD)。结果表明,各基质中3种目标化合物的回收率为66.6%~120%,RSD为0.80%~11%(表2),能够达到检测痕量残留目标物的要求。

表2 目标化合物的检出限、定量下限、回收率及相对标准偏差(n=5)Table 2 Detection limits,quantitative limits,recoveries and relative standard deviations of the target analytes(n=5)
2.8 实际样品检测
按本法对实验室历年收集的不同厂家共45批样品进行测定,其中10批样品检出氯霉素,检出率为22.22%,残留量为0.14~14.32 μg/kg;8批样品检出氟甲砜霉素,检出率为17.78%,残留量为0.22~21.62 μg/kg(见表3),说明蜂胶及其原料保健食品中存在较大的氯霉素、氟甲砜霉素残留风险。代表性阳性样品的MRM谱图见图2。
图2 同时检出氯霉素和氟甲砜霉素的阳性样品的多反应监测(MRM)谱图Fig.2 MRM chromatograms of simultaneous detection of chloramphenicol and florfenicol positive samples

表3 阳性样品的检测结果Table 3 Determination results of positive samples
3 结 论
本文根据蜂胶原料及蜂胶原料保健食品的基质特点,建立了同时检测氯霉素、甲砜霉素、氟甲砜霉素残留的HPLC-MS/MS法,并对市售样品进行了检测。结果同时检出了氯霉素及氟甲砜霉素,两者的最大残留量分别为14.32、21.62 μg/kg,其中氟甲砜霉素阳性检出为国内外首次报道,说明关注蜂胶原料及蜂胶原料保健食品中的兽药残留是必要的。本方法操作简单,灵敏度高,重复性好,应用前景良好,可满足实际检验工作的需要。
