超高效液相色谱-串联质谱法同时测定化妆品中8种双酚类及烷基酚类内分泌干扰物
2020-08-03袁晓倩简龙海毛北萍
袁晓倩,韩 晶,简龙海,陈 静,毛北萍,郑 荣
(上海市食品药品检验所,国家药品监督管理局化妆品监测评价重点实验室,上海 201203)
双酚类和烷基酚类化合物是生产聚碳酸酯塑料和环氧树脂的重要原料,被广泛应用于食品、化妆品包装材料、医疗器械、工业及家用洗涤剂等领域[1-4]。作为典型的雌激素类环境内分泌干扰物,这两类化合物具有不易降解和易蓄积的特性,从空气到土壤均有被污染的风险[5]。内分泌干扰物可以通过食物链进入机体,并在体内富集,对生物体具有基因毒性、生殖发育毒性和内分泌干扰效应,并且对婴幼儿的危害尤为严重[6-8]。我国《化妆品安全技术规范》(2015年版)和欧盟化妆品法规(EC No 1223/2009)明确规定双酚A、壬基苯酚与支链4-壬基苯酚为化妆品中的禁用物质,但目前尚未收载相关检验方法[9-10]。因此,迫切需要开发同时测定多种双酚类及烷基酚类内分泌干扰物的检测方法,并应用建立的分析方法对化妆品样品进行全面调研。
目前,国内外关于双酚A、4-壬基苯酚和4-辛基苯酚的检测方法主要有电化学法[11-12]、高效液相色谱法[13-15]、分光光度法[16]、毛细管电泳法[17]、酶联免疫吸附分析法[18]、气相色谱-质谱法[19-20]和液相色谱-串联质谱法[21-26]等。但光谱法、色谱法存在选择性差、灵敏度低等不足,无法解决化妆品基质复杂的问题,阳性样品需经质谱进一步确证。而现有的超高效液相色谱-串联质谱法响应低,需进一步优化色谱与质谱条件。此外,目前已有文献报导的检测方法覆盖化合物较少,仅限于1~2种化妆品基质。
本研究采用超高效液相色谱-三重四极杆质谱法对化妆品中的双酚类化合物(双酚A、双酚S、双酚F)和烷基酚类化合物(对特辛基苯酚、2,4-二叔丁基酚、支链4-壬基苯酚、4-辛基苯酚、4-壬基苯酚)进行检测。建立的方法快速灵敏、分离度好,能满足化妆品中相关物质的检测要求,从而为化妆品的日常监督和风险评估提供了技术支持。
1 实验部分
1.1 仪器与试剂
超高效液相色谱仪(美国Agilent公司);API 6500型串联四极杆质谱仪(美国Absciex公司);分析天平(德国Sartorius公司);MS 3 digital涡旋混合器(德国IKA公司);5810 R离心机(德国Eppendorf公司);Milli-Q Advantage A-10型超纯水纯化系统(美国Millipore公司)。
双酚S(纯度99.30%)、双酚F(纯度99.81%)、双酚A(纯度99.60%)、对特辛基苯酚(纯度98.65%)、4-辛基苯酚(纯度99.28%)、4-壬基苯酚(纯度99.72%),均购自德国Dr.Ehrenstorfer公司;2,4-二叔丁基酚(纯度99.80%,德国CNW公司);支链4-壬基苯酚(纯度99.80%,美国o2si标准品公司);双酚A内标(纯度99.00%,美国CDN标准品公司);甲醇、乙腈(色谱级,Merck公司);氨水(分析纯,上海凌峰化学试剂有限公司)。实验用水由Milli-Q超纯水机制备。
1.2 实验条件
1.2.1 色谱条件色谱柱:Waters X-Bridge BEH Phenyl液相色谱柱(2.1 mm×100 mm,2.5 μm);柱温:25 ℃;流速:0.3 mL/min;进样体积:5 μL;流动相:0.1% 氨水(A)和甲醇(B),梯度洗脱程序:0~3 min,70%~40% A;3~10 min,40%~10% A;10~15 min,10% A;15~15.1 min,10%~70%A;15.1~18 min,70%A。
1.2.2 质谱条件离子化模式:负离子电喷雾离子化(ESI-);质谱扫描方式:多反应监测(MRM);气帘气压力(CUR):1.72×105Pa,氮气;离子源喷雾电压(IS):5 500 V;雾化温度(TEM):450 ℃;雾化气压力(GAS1):55 psi(3.79×105Pa),氮气;加热辅助气(GAS2):50 psi(3.45×105Pa),氮气;离子驻留时间均为50 ms;各化合物的检测离子对、去簇电压(DP)、碰撞能量(CE)见表1。
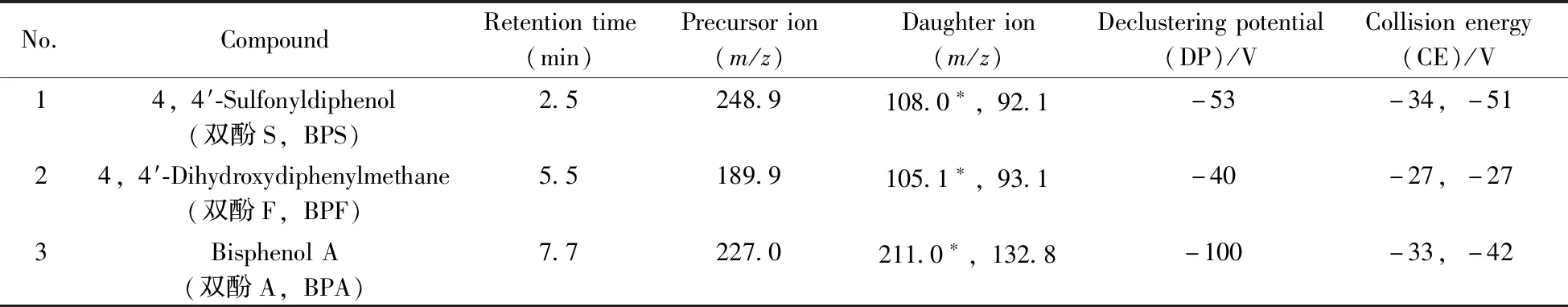
表1 9种目标分析物的监测离子对及相关电压参数Table 1 Monitor ion pairs and voltage parameters of 9 target analytes
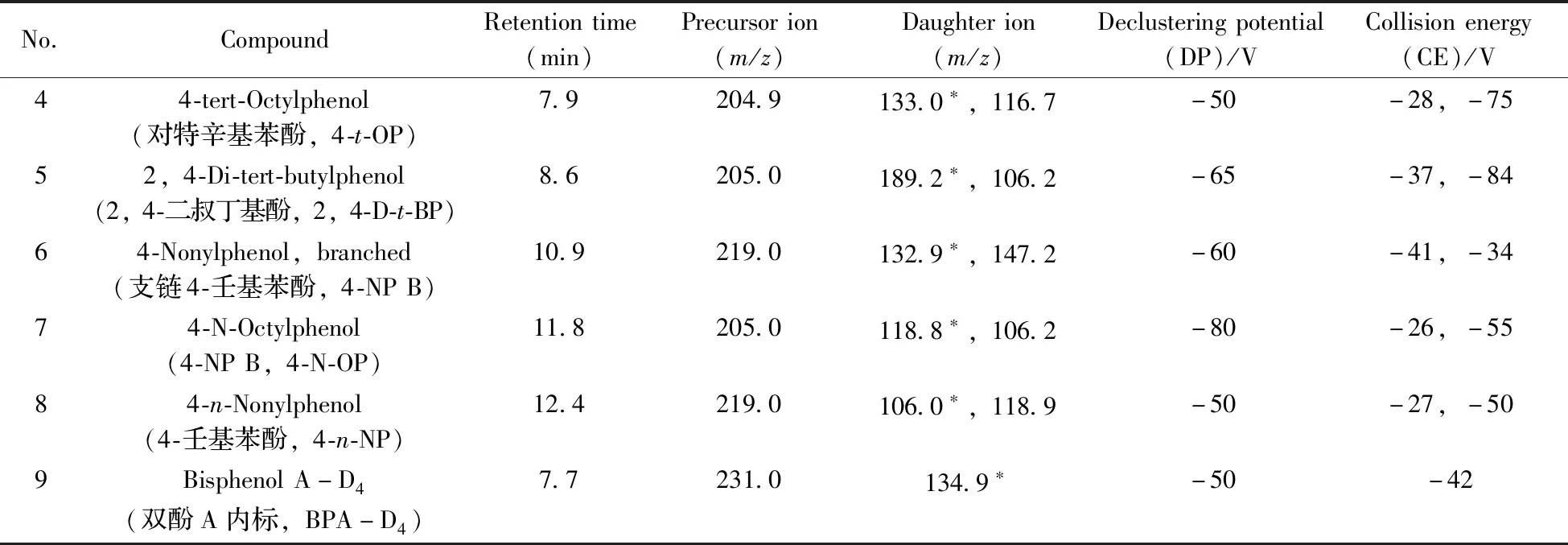
(续表1)
1.3 标准溶液的配制
精密称取双酚A内标标准品10 mg置于10 mL量瓶中,用甲醇溶解并定容至刻度,精密吸取0.1 mL置于100 mL量瓶中,用甲醇稀释至刻度,配制成质量浓度为1 μg/mL的同位素内标工作溶液。
分别精密称取8种待测物标准品10 mg置于10 mL量瓶中,用甲醇溶解并定容至刻度,制成质量浓度为1 mg/mL的混合标准储备溶液,于4 ℃冰箱保存。
分别精密吸取上述混合标准品溶液和同位素内标工作溶液适量,用甲醇稀释成8种标准品质量浓度为0.005、0.2、0.5、1、2、5、10、20、50 ng/mL的系列待测物标准工作溶液,内标溶液质量浓度均为10 ng/mL。
1.4 样品前处理
粉类、水剂、膏霜乳液类、凝胶类化妆品:准确称取化妆品样品约1.0 g,精确至0.001 g,置于25 mL玻璃比色管中,加入250 μL双酚A内标工作溶液(1 μg/mL),再加入乙腈-1%氨水(4∶1,体积比)混合溶液定容至刻度,涡旋混匀,超声提取30 min,4 500 r/min离心5 min,取上清液经0.45 μm微孔滤膜过滤后,滤液作为待测液备用。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 色谱柱的选择由于待测的烷基酚类化合物中包含同分异构体,其具有相同的母离子和子离子,因此需选择合适的液相色谱柱将其完全分离。本实验尝试了3种柱效较高的快速分离色谱柱:A柱为Agilent Poroshell 120 EC C18(4.6 mm×100 mm,2.7 μm),B柱为Agilent ZORBAX Eclipse Plus C18(2.1 mm×100 mm,1.8 μm),C柱为Waters X-Bridge BEH Phenyl(2.1 mm×100 mm,2.5 μm)。实验发现,A柱可有效分离双酚类化合物,但对烷基酚类化合物无保留。B柱可分离部分烷基酚类化合物,但对双酚类化合物的保留较差。C柱可以同时有效分离双酚类及烷基酚类化合物,且峰形最优(如图1所示)。因此,实验选择Waters X-Bridge BEH Phenyl(2.1 mm×100 mm,2.5 μm)为最佳色谱柱。
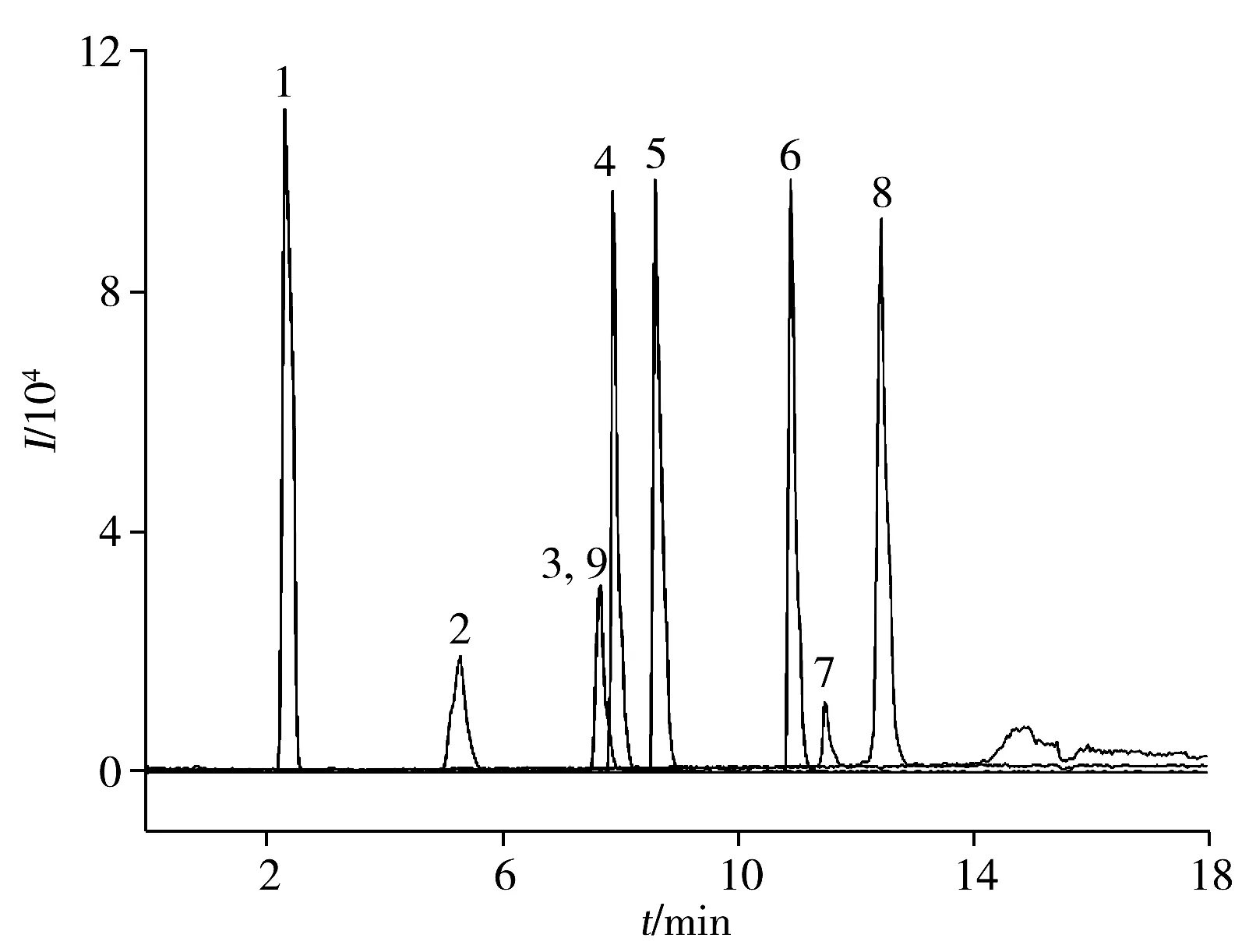
图1 9种目标分析物的MRM色谱图Fig.1 MRM chromatogram of 9 target analytes the number denoted was the same as that in Table 1
2.1.2 流动相的选择由于酚类化合物多在ESI-模式下离子化,而甲醇相比于乙腈具有更低的表面张力,有利于电喷雾液滴源内溶剂挥发,提高灵敏度。本实验以甲醇为有机相,考察了0.1%氨水、1%氨水、5 mmol/L乙酸铵、纯水作为水相的响应,发现前三者均能够提高离子对响应。但随着氨水浓度的增加,双酚F出现色谱峰拖尾的情况。综合考虑峰形与离子对响应,选用0.1%氨水和甲醇作为最佳流动相体系。
2.2 前处理条件的优化
根据目标化合物的极性,本实验考察了1%氨水甲醇、1%氨水乙腈、乙腈-1%氨水(4∶1,体积比)、乙腈-1%氨水(1∶1,体积比)4种溶剂对化妆品基质的提取效果。结果表明,粉类、水剂、膏霜乳液类、凝胶类4种基质的化妆品均能均匀分散在4种溶剂中,但使用乙腈-1%氨水(1∶1)提取时,待测物的回收率仅为67.5%~79.2%。使用1%氨水甲醇提取时,长链烷基酚(4-NP B,4-n-NP,4-N-OP)的回收率为63.4%~75.1%。使用1%氨水乙腈提取时,回收率虽能达到91.5%~98.4%,但由于1%氨水乙腈的溶剂效应,易造成峰形拖尾。而选择乙腈-1%氨水(4∶1)为提取溶剂时,可在确保回收率(82.2%~99.1%)的同时(见图2),保证峰形尖锐对称、分离度好。故本实验选择乙腈-1%氨水(4∶1)对8种待测物进行提取。
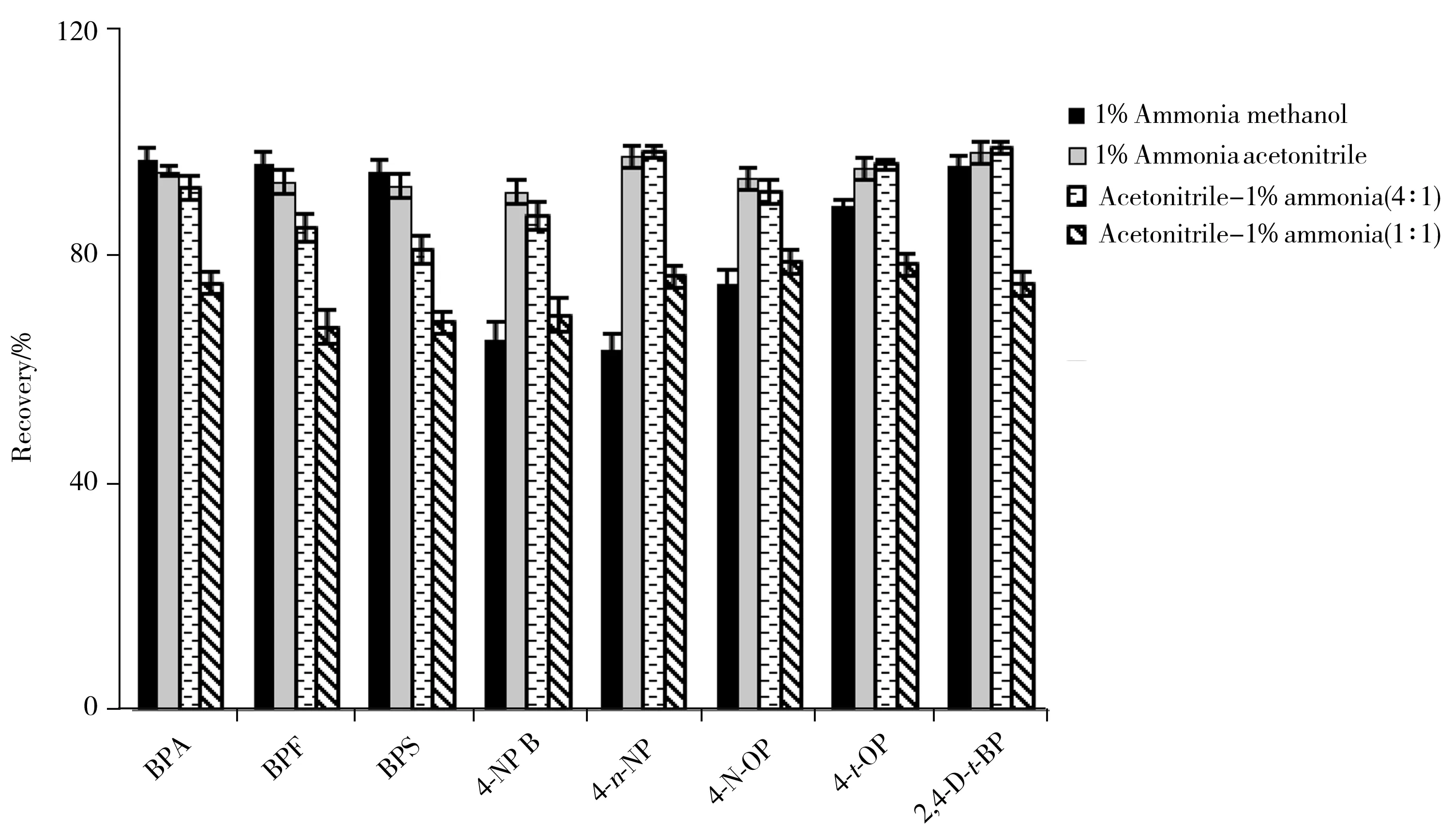
图2 4种提取溶剂对膏霜类样品中8种待测物的提取效果Fig.2 Extraction efficiencies of 8 analytes in cream samples by four solvents
2.3 方法学验证
2.3.1 线性关系、检出限与定量下限在优化条件下,双酚A采用内标法,其他7种双酚类及烷基酚类化合物采用外标法对“1.3”配制的系列浓度(0.005~50 ng/mL)标准工作溶液进行测定,以各组分的质量浓度(X,ng/mL)为横坐标,双酚A的峰面积与内标峰面积的比值(其他7种待测物的峰面积)(Y)为纵坐标,绘制标准曲线。由表2可知,8种内分泌干扰物均在一定质量浓度范围内线性关系良好,相关系数(r)大于0.997。
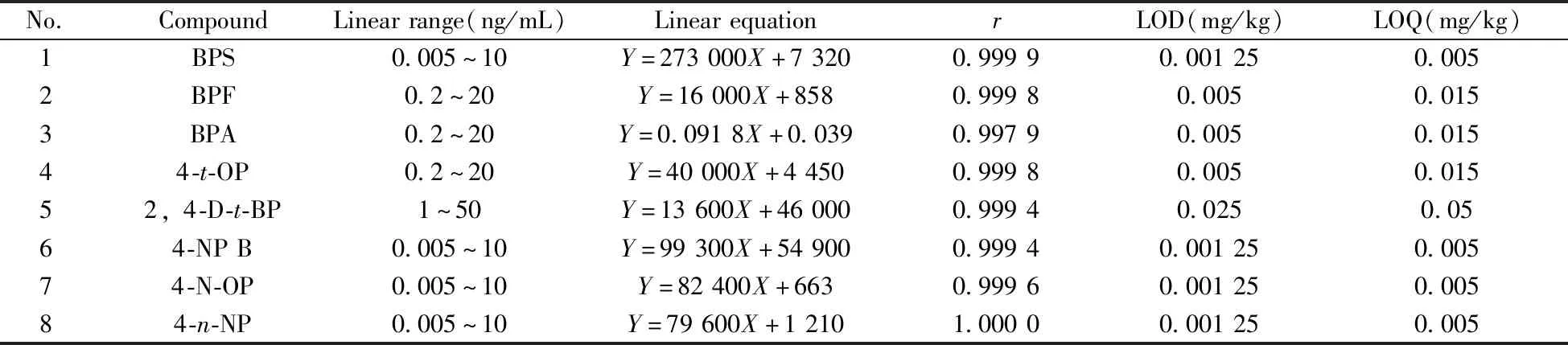
表2 8种目标分析物的线性范围、线性方程、相关系数、检出限及定量下限Table 2 Linear ranges,linear equations,correlation coefficients(r),limits of detection(LOD) and limits of quantitation(LOQ)of 8 target analytes
选取粉类、水剂、膏霜乳液类、凝胶类阴性样品,各称取1.0 g,添加混合标准储备溶液适量,按本方法进行前处理并测定。分别以信噪比(S/N)为3和10时进行计算,得到方法的检出限(LOD)为0.001 25~0.025 mg/kg,定量下限(LOQ)为0.005~0.05 mg/kg(见表2)。其中,本方法对双酚A的检出限(0.005 mg/kg)低于现有国标方法GB/T 30939-2014的检出限(0.025 mg/kg)[27]。
2.3.2 加标回收率与相对标准偏差取粉类、水剂、膏霜乳液类、凝胶类阴性样品各1.0 g,平行6份,分别加入不同体积的混合标准储备溶液,按照本方法进行前处理并测定,方法的加标回收率和相对标准偏差(RSD)见表3。由表3可知,8种内分泌干扰物的回收率为75.1%~107%,RSD(n=6)为0.1%~1.6%,表明本方法的准确度和精密度较高。
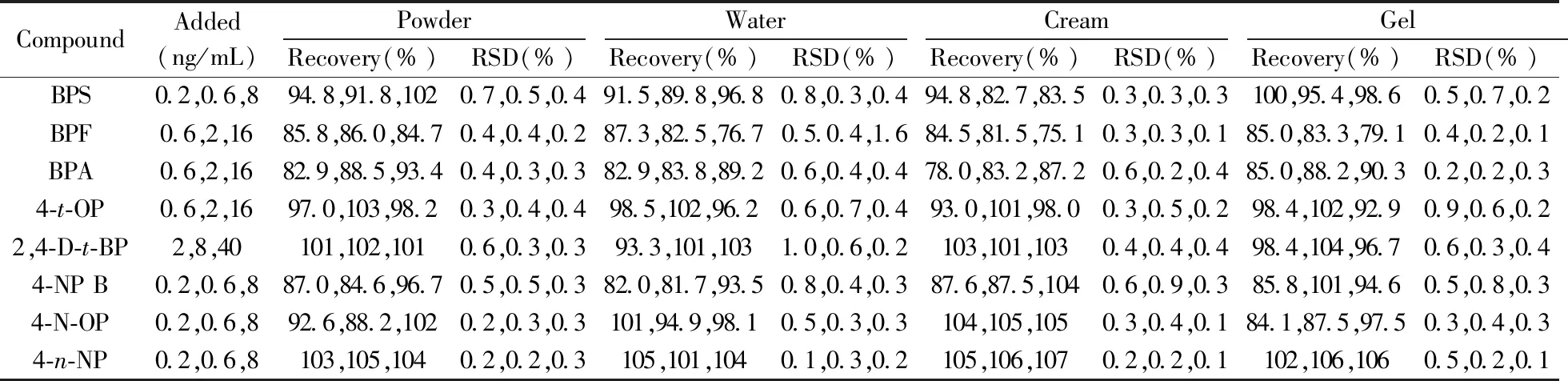
表3 回收率与重复性实验结果Table 3 Results of spiked recovery and repeatability test
2.4 实际样品的测定
应用本方法分别对从市场收集的不同场地、不同品牌的61批染发类样品进行测定,其中双酚A检出5批次,含量为0.017~0.475 mg/kg;支链4-壬基苯酚检出14批次,含量为0.020~1.190 mg/kg;2,4-二叔丁基酚检出2批次,含量为0.236~0.250 mg/kg。实际样品检测结果表明,有必要进一步加强对染发类样品的监管力度。
针对阳性样品,采用国标方法GB/T 30939-2014与本方法进行对比。由于仅双酚A有国标检测方法(检出限0.025 mg/kg),故仅对双酚A作方法比较。结果显示,以某染发霜为典型样品,采用本方法测得双酚A的含量为0.475 mg/kg,样品的MRM提取色谱图如图3所示,国标方法测得该染发霜中双酚A含量为0.465 mg/kg。本方法与国标方法的结果基本一致。

图3 阳性样品的提取离子色谱图Fig.3 Extracted ion chromatogram of a positive sample
3 结 论
本文建立了UPLC-MS/MS同时测定化妆品中8种双酚类及烷基酚类化合物的分析方法。该方法使用内标法定量,样品前处理简单、回收率稳定、灵敏度高,适用于多种化妆品基质中内分泌干扰物的定性定量分析。该方法的建立进一步拓展了双酚A等内分泌干扰物的分析技术,可为化妆品监管提供有力的技术支撑。
