溪黄草转录组测序及生物信息学分析
2020-07-24王继华蔡时可梅瑜李汉章杨少海
王继华,蔡时可,梅瑜,李汉章,杨少海
(1.广东省农作遗传改良重点实验室,广东省农业科学院作物研究所,广东广州 510640;2.广东建鹏南药种养有限公司,广东广州 510070)
溪黄草Rabdosia serra(Maxim.)Hara 为唇形科香茶菜属多年生草本植物,喜阴凉湿润环境,俗称溪沟草、黄汁草等,多产于华南地区的广东、广西及台湾[1-2]。全草均可入药,味苦、性寒,具有清热利湿、退黄、凉血散瘀的功效,用于治疗湿热泻痢、跌打瘀肿、急性黄疸型肝炎、急性胆囊炎、口腔炎、肠炎等病证[3-5]。药理学研究表明,溪黄草具有保肝、护肝、抗炎、抗癌作用,是多种中成药和保健品的主要成分[3,6-7]。溪黄草是广东省连州市的特产,连州溪黄草在2007 年获得国家地理标志产品(国家质量监督检验检疫总局,2007年第220号公告)。
溪黄草来源复杂,其基原植物为唇形科溪黄草、线纹香茶菜、以及线纹香茶菜的变种细花线纹香茶菜和狭基线纹香茶菜[8]。资源鉴定主要根据根、茎、叶、花、果实等形态鉴定及味道等进行区分[9-11]。溪黄草中主要含有萜类、黄酮类、多糖和酚酸类等化学成分,各种有效成分还具有协同增效作用[12]。不同的基源植物之间化学成分相近,但部分药效成分的含量相差甚远[13]。溪黄草基因组数据仍不完整,不能对有效成分的代谢途径进行深入的挖掘。目前,高通量测序平台的RNA-seq技术在解析药用成分代谢通路、挖掘关键基因、开发分子遗传标记等方面得到广泛应用[14-16]。因此,本研究开展溪黄草的转录组测序及生物信息学分析,以期为解析其药用物质合成代谢通路、挖掘关键调控基因以及开发分子标记奠定基础。现将研究结果报道如下。
1 材料与方法
1.1实验材料实验材料为广东省农业科学院作物研究所南药资源圃栽培的溪黄草。于2018 年12 月份取样,采集健壮植株的叶片,迅速用锡箔纸包裹并浸入液氮处理,随后置于超-80 ℃冰箱保存。
1.2溪黄草RNA的提取采用生工生物工程(上海)股份有限公司的总RNA 提取试剂盒(B511311-0025)提取溪黄草总RNA,然后通过1%电泳凝胶检测提取RNA的完整性。应用Invitrogen Qubit®2.0荧光计及试剂盒(Fluorometer Life Tech Invitrogen,Q32886)对总RNA进行定量。
1.3转录组测序与拼接组装委托生工生物工程(上海)股份有限公司采用Illumina HiSeq2500 的高通量测序平台进行转录组测序。测序得到的原始数据,通过FastQC软件进行质量评估和Trimmomatic进行质量剪切,过滤掉接头、低质量的序列(reads < 35nt)、带N 碱基的序列、低质碱基(Q值<20)得到高质量的clean data[17]。应用Trinily 软件对clean data 进行de novo拼接组装,再采用RSeQC软件去除转录本中的冗余序列,得到非冗余通用基因(universal gene,unigene)[18]。
1.4基因功能注释采用基于局部比对算法的搜索工具(Basic Local Alignment Search Tool,BLAST)将组装的unigene 与保守域数据库(Conserved Domain Database,CDD)、真核生物蛋白质同源簇数据库/蛋白相邻的聚类(eu Karyotic ortholog groups/clusters of orthologous groups,KOG/COG)、非冗余(Non-redundant,NR)、核酸序列数据库(Nucleotide Sequence Database,NT)、蛋白结构域预测(Protein Families Database of Alignments and Hidden Markov Models,PFAM)、Swissprot、TrEMBL 等多个数据库比对得到功能注释信息。使用Transdecoder进行编码序列(Coding Sequence,CDS)预测。根据unigene 与Swissprot、TrEMBL 的注释结果得到基因本体论(Gene Ontology,GO)功能注释信息,利用京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)自动注释服务器(KEGG Automatic Annotation Server,KAAS)得到KEGG注释信息。
1.5基因结构分析使用微卫星识别工具(Microsatellite Identification Tool,MISA)软件鉴定溪黄草unigene上存在的SSR 位点,并利用Primer 3软件(http://primer3.sourceforge.net/releases.php)设计SSR引物[19]。
2 结果与分析
2.1转录组测序与de novo组装溪黄草cDNA 文库的构建由生工生物工程(上海)股份有限公司完成。应用Illumina Hiseq 2500测序平台测序,共获得61 944 850条raw reads,总碱基数为9 291 727 500 bp。使用Trimmomatic 对原始测序数据进行处理,去掉含有带接头、低质量的序列,共得到60 234 786 clean reads,总碱基数目为8 704 764 735 bp,GC含量为51.21%,Q30 bases ratio达到95.64%,表明文库构件质量良好,测序得到的数据准确可靠。使用Trinity 将clean reads 进行de novo组装成转录本,共得到86 204 条转录本,平均长度为1 251.93 bp,N50为1 894 bp,序列长度大于500 bp的有59 403条,占总序列数目的68.91%。见表1。对Trinity拼装得到的转录本去冗余,共获得37 418 条unigene,平均长度为1 054.1 bp,N50为1 840 bp,其中20 426条序列长度大于500 bp,占总序列数目的54.59%,序列长度在1 000 bp以上的有14 226条,占总数的38.02%见。见表1、图1。

表1 溪黄草转录组测序的结果Table 1 Summary of assembled transcripts and unigenes of the Rabdosia serra(Maxim.)Hara transcriptome

图1 溪黄草unigene序列长度分布Figure 1 Length distribution of assembled unigenes of Rabdosia serra(Maxim.)Hara
2.2 Unigene功能注释将溪黄草组装后的unigene序 列 与 CDD、 KOG、 COG、 NR、 NT、 PFAM、Swissprot、TrEMBL 等多个数据库进行比对,共有23 978 条(64.08%)unigene 在至少一个数据库中获得功能注释,2 429 条(6.49%)unigene 在所有数据库种均能获得注释,见表2。其中NR 数据库中注释到的unigene 数目(23 623 条)最多,占总unigene的63.13%,其次为TrEMBL 数据库,注释到的unigene 为 23 393 条,占 62.52%。KEGG 数据库中注释到的unigene 数目(3 674 条,9.82%)最少。尚有13 440条unigene没有得到有效注释,占35.92%。见表2。通过与NR 库的比对,获得溪黄草unigene序列与近缘种属的近似情况并获得同源序列的功能信息,共有23 616条unigene获得注释。匹配较多的物种主要有Sesamum indicum、Erythranthe guttata、Dorcoceras hygrometricum和Salvia miltiorrhiza,分别占58.88%、20.39%、3.16%和1.97%,见图2。表明溪黄草与唇形目(管状花目)Sesamum indicum的序列相似度最高。
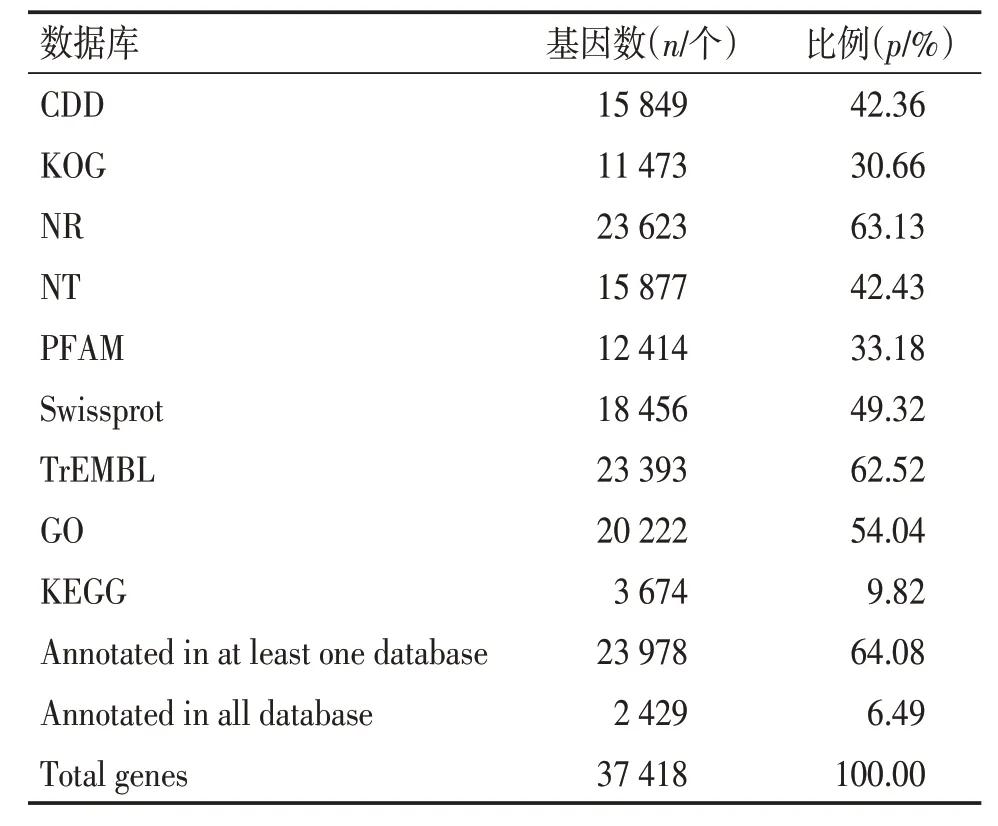
表2 Unigene 的功能注释Table 2 Functional annotation of assembled unigenes
2.3 KOG功能注释将组装得到的溪黄草unigene与KOG数据库比对,共有11 473条unigene获得注释,按其功能共分为26 类。其中:信号转导机制注释到的unigene 数目最多,共有1 974 条,占15.35%;一般功能预测、翻译后修饰,蛋白质转运,分子伴侣伴侣、转录和碳水化合物的转运和代谢注释到的unigene 占比分别为11.42%、10.26%、5.50%和5.21%;仅有34条和4条unigene注释对应到核结构和细胞运动性,分别占比0.26%和0.03%。见图3。此外,676 条unigene 注释到未知功能。由此可见,溪黄草unigene 涉及信号转导功能最多,可为今后溪黄草代谢物质的调控研究提供宝贵资源。

图2 NR数据库的同源物种分类Figure 2 Species distribution of the top BLAST hits against the NR database for the assembled unigenes
2.4 GO功能注释GO 数据库是全面描述生物体中基因及其产物属性的分类系统,主要分为生物过 程(biological process)、 细 胞 组 分(cellular component)及分子功能(molecular funtion)三大类,见图4。根据GO 数据库注释结果,共有20 222 条溪黄草unigene注释成功。这些unigene总共被划分为65 个功能分类,生物过程注释到的unigene 最多,共有42 872 个,占35.95%,其中注释较多的功能分别为细胞过程(11 757个,7.88%)、代谢过程(9 935个,6.66%)、刺激响应(5 455个,3.66%)和生物调节(4 952 个,3.32%)。注释到分子功能的unigene 数目为38 425个,占32.22%,其中注释较多的功能分别为结合(11 625 个,7.79%)、催化活性(9 633,6.45%)和转运活性(1 276个,0.86%)。37 943 个(31.82%)unigene 注释到细胞组分,其中注释较多的功能分别为细胞(14 297 个,9.58%)、细胞组分(14 265 个,9.56%)、细胞器(10 568 个,7.08%)、膜(8 331 个,5.58%)和膜组分(6 481 个,4.34%)。

图3 溪黄草unigene的KOG功能分类Figure 3 KOG function classification of Rabdosia serra(Maxim.)Hara unigenes

图4 溪黄草unigene的GO功能分类Figure 4 GO classification of Rabdosia serra(Maxim.)Hara unigenes
2.5 KEGG功能注释根据KEGG 数据库注释结果,主要分为五大类功能,包括代谢(2 529 个,41.67%)、遗传信息过程(1 298 个,21.39%)、细胞过程(638 个,10.51%)、环境信息过程(623 个,10.27%)和有机系统(981 个,16.16%)。根据unigene 参与的代谢过程,进一步划分为32个功能分类,涉及289 个代谢通路,具体见图5。在代谢中,与氨基酸代谢和脂质代谢相关的unigene 分别为305 个(5.03%)和275 个(4.53%)。在有机系统中,涉及环境适应的unigene 有162 个,占2.67%。在遗传信息过程中注释到unigene 最多的代谢过程为转录(212 个,3.49%),而在细胞过程中,与转运和分解代谢的unigene 最多(310个,5.11%)。信号转导在植物发育过程以及对外界刺激的响应过程起重要作用,本研究共检测到590 条unigene 涉及信号转导,可为下一步鉴定溪黄草的信号因子提供研究基础。溪黄草主要含萜类、黄酮类、酚类、氨基酸等化学成分,其中二萜类化合物非常丰富。本研究鉴定到19个unigene涉及黄酮类物质的生物合成,12个unigene 涉及倍半萜类化合物和三萜类化合物生物合成,40个unigene 与萜类物质骨架生物合成相关,13个unigene 参与双萜的生物合成,51个unigene 涉及苯丙素的生物合成。这些与次生代谢相关unigene 的鉴定结果为进一步解析溪黄草药用成分物质的生物合成提供了可能。

图5 溪黄草unigene的KEGG功能分类Figure 5 KEGG classification of Rabdosia serra(Maxim.)Hara unigenes
2.6 SSR分析见图6。采用MISA 对组装的unigene 进行SSR 检测,并对SSR 的类型和密度进行统计。结果表明,在7 809条unigene中共鉴定到9 489 个 SSR 位点。其中,921 条 unigene 中检测到944 个(9.95%)复杂重复类型的SSR 位点。最丰富的重复类型是双碱基重复,共检测到4 208 个位点,占44.35%;其次为单碱基重复(2 524 个,26.60%)、三碱基重复(1 685 个,17.76%)、六碱基重复(70 个,0.74%)和四碱基重复(42 个,0.44%);最少的为五碱基重复,仅检测到16 个位点,占0.17%。见图6-A。在SSR 分布的密度上,双碱基重复最高,达到142.2个/Mbp,五碱基重复最低,仅为0.48 个/Mbp。见图6-B。基于9 489 个SSR 位点,使用Primer 3.0 设计引物,为进一步开发溪黄草的遗传标记和近缘种属的遗传图谱提供了基础数据。

图6 溪黄草unigene的SSR位点分析Figure 6 The analysis of SSR sites of Rabdosia serra unigenes
3 讨论
近年来,随着高通量测序技术的发展,凭借其产出数据量大、成本低、不需要参考基因组等优势,在新基因发掘、功能基因鉴定和分子标记开发上的应用越来越广泛。本研究基于Illumina HiSeq2500的高通量测序平台对溪黄草叶片进行转录组测序,共获得60 234 786 条clean reads,总碱基数目为8 704 764 735 bp,GC 含量为51.21%,Q30 bases ratio 达到95.64%,表明文库构件质量良好。N50 是评价组装序列完整性的重要指标。通过de nove组装,本研究共获得37 418 条unigene,平均长度为1 054.1 bp,N50为1 840 bp,与已经构建云南松(1 818 bp)、香榧(1 702 bp)的N50 长度相接近,但较中药黄芩(797.64 bp)的N50 长,表明溪黄草转录组序列组装质量较高[20-22]。
溪黄草组装后的unigene 序列与多个公共数据库比对,共有 23 978 条(64.08%)unigene 在至少1 个数据库中获得功能注释,2 429 条(6.49%)unigene在所有数据库中均能获得注释,但仍有约35.92%的unigene 没有获得注释信息,可能是由于部分序列组装长度过短,缺少保守的核心序列以及溪黄草基因组信息匮乏暂时无法获得准确的功能注释。在KOG 数据库和KEGG 数据库中分别鉴定到1 974条和590条unigene涉及信号转导,可为下一步解析溪黄草响应外界刺激以及代谢物质的调控提供靶标基因。同时,还鉴定到19个unigene涉及黄酮类物质的合成,51个unigene 涉及苯丙素的生物合成,以及大量与萜类物质合成相关的unigene,这为明晰溪黄草黄酮类和萜类物质的合成途径和代谢网络提供了数据支撑。
SSR标记操作简单、重复性好,与传统方法相比,高通量能够挖掘出大量的SSR 位点。甘草、铁皮石斛、枸杞等中药材均利用转录组测序开发了SSR 标记用于分子辅助育种和遗传图谱的构建[23-25]。本研究应用MISA 软件共检测到9 489 个SSR 位点,最丰富的重复类型是双碱基重复(4 208 个位点),占44.35%,达到142.2 个/Mbp,该研究结果与中药黄芩类似[20]。本研究对溪黄草转录组进行了初步的探究,弥补了溪黄草基因组信息的不足,为解析次级代谢物质合成通路及分子生物学方面的研究打下了基础。溪黄草SSR 位点的发掘,可为溪黄草分子标记的开发、遗传多样性分析、遗传图谱构建等奠定理论基础,为利用分子手段鉴定和区分溪黄草及其基源植物提供了依据。
