一种二苯乙烯羟胺类化合物的合成及其在糖类检测中的应用研究
2020-07-15赵孔娥朱芳芳胡晓松
赵孔娥, 朱芳芳, 马 傲, 朱 灏, 胡晓松
(武汉理工大学 化学化工与生命科学学院,湖北 武汉 430070)
糖基化是修饰蛋白质的重要方式[1]。修饰后的蛋白质可参与到人体的各项生命活动中,如细胞识别、细胞免疫、信息传递等[2]。糖基化异常与很多疾病有关[2-3]。因此针对糖蛋白的聚糖进行监测分析对疾病的诊断与治疗有重要意义。然而,由于糖类物质结构复杂、离子化效率较低,也不具备生色团,用常规的有机波谱分析方法开展其结构研究十分困难[4]。在分析聚糖前通常需要进行衍生化处理。近来,结合稳定的同位素标记的质谱分析成为定量糖组学研究中最可行的策略之一。对化学上相似的聚糖采用含“轻”和“重”的稳定同位素的衍生试剂进行衍生,不仅能有效消除电离效率的差异,还可以同时对多个样品进行质谱分析,“轻”和“重”同位素标记的聚糖衍生物质谱峰具有明显的质荷比区别;通过比较它们的相对强度,可以获得具有相同化学特征的聚糖的相对含量。常见的用于聚糖定量分析的衍生方法包括全甲基化法[11],还原胺化法[12-14]和腙化法[15]。所有这些定量策略都是通过用H和D(或12C和13C)修饰的同位素标记试剂和聚糖反应来实现的。
羟胺类化合物由于具有很高的亲和性及其它特定的性质,在化学合成、药物化学以及化学生物学方面得到了广泛的应用[16]。O-取代和N-取代羟胺类衍生物已经在醛类化合物的检测分析中取得了一系列成果[4,17-18],也可以用于聚糖分子的衍生分析。在大多数情况下,O-取代羟胺类衍生化反应通过氨氧基与醛基反应形成肟,然后进一步被还原后进行定量分析。而N-取代的羟胺类衍生化反应是通过-NHOH与醛基反应形成硝酮类化合物,直接用于定量分析[17],也可以进一步被还原(Chart 1)。二苯乙烯的π共轭有机体系具有较高的光学可调控性和拓展性[19, 20],是一个良好的荧光基团。二苯乙烯片段可以通过苯乙烯参与的Heck反应制得,如果选择全氘代苯乙烯(styrene-d8)为原料,则可以制得氘标记的重原子同位素探针。基于这些分析,我们设计、合成了二苯乙烯类羟胺化合物(5a和5b, Scheme 1),并初步用于糖类分子的检测。
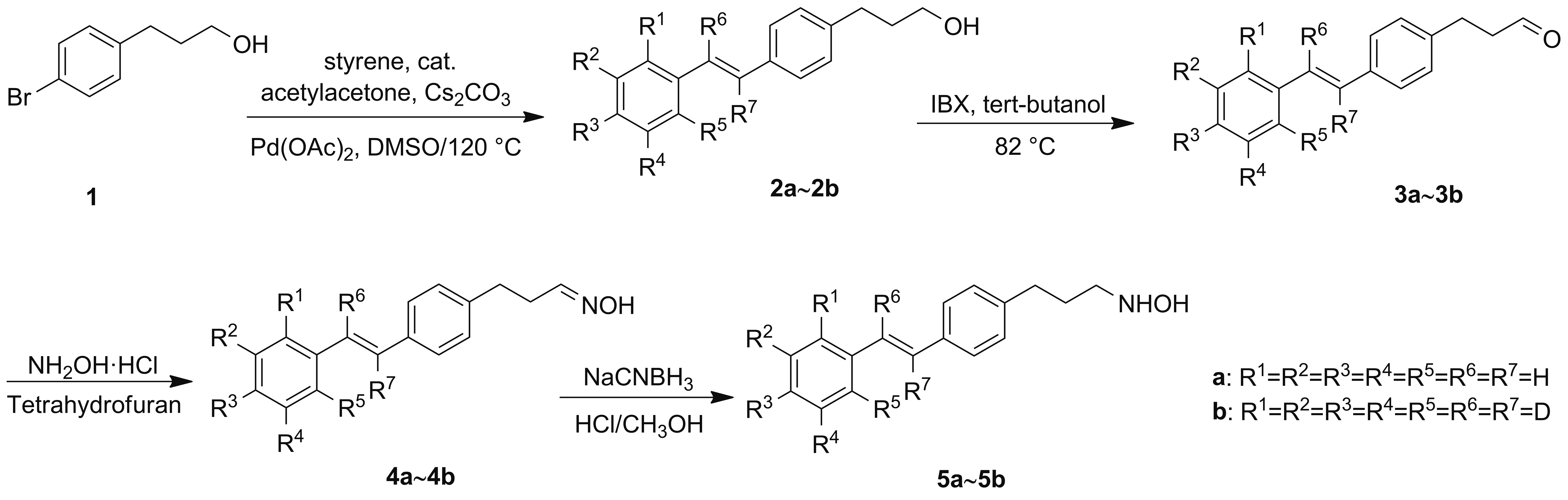
本文按照Scheme 1的合成路线合成了二苯乙烯类羟胺化合物:先以4-溴苯丙醇(1)和苯乙烯为原料,在醋酸钯的催化下通过Heck反应得到(E)-3-(4-苯乙烯基苯基)丙烷-1-醇(2a);2a被2-碘酰基苯甲酸(IBX)氧化为(E)-3-(4-苯乙烯基苯基)丙醛(3a);3a再与盐酸羟胺反应得到(E)-3-(4-苯乙烯基苯基)丙醛肟(4a);丙醛肟4a在酸性条件下被氰基硼氢化钠还原为(E)-N-(3-(4-苯乙烯基苯基)丙基)羟胺(5a),总产率为4.8%。采用相同的路线,以全氘代苯乙烯为起始原料,合成了(E)-N-(3-(4-(2-(苯基-d5)乙烯基-1,2-d2)苯基)丙基)羟胺(5b)。化合物5a和5b可以分别作为“轻”和“重”的稳定同位素衍生试剂。化合物结构经1H NMR,13C NMR, IR和LC-MS(ESI)表征。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪;UV757CRT型紫外-可见分光光度计;Bruker AVANCE III 500 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Nicolet iS5型傅里叶红外光谱仪(KBr压片);Triple TOFTM5600型质谱仪;G2-XS QTof型质谱仪;LS55型荧光/磷光/发光分光光度计;ZF-7A型手提式紫外分析仪。
4-溴苯丙醇(Adamas);苯乙烯(Alfa Aesar); 2-碘酰基苯甲酸(IBX)(Innochem);叔丁醇(Aladdin);盐酸羟胺(阿拉丁);氰基硼氢化钠(Innochem);N,N-二异丙基乙胺(上海秦巴化工有限公司);三苯基膦(国药集团化学试剂有限公司);麦芽三糖(DP3)、麦芽五糖(DP5)和麦芽七糖(DP7)(西宝生物科技);其余所用试剂均为化学纯或分析纯。
1.2 合成
(1)2a的合成
将化合物10.22 g(1.00 mmol), 10 mol%Pd(OAc)222.45 mg(0.10 mmol), 20 mol%乙酰丙酮20.01 mg(0.2 mmol), 10 mol%四丁基溴化铵(TBAB)32.24 mg(0.1 mmol), Cs2CO3651.64 mg加入DMSO(2 mL)中,氮气保护下,加入苯乙烯312.45 mg(3.00 mmol),于120 ℃反应2 h。冷却至室温,用EtOAc(100 mL)稀释,依次用饱和氯化铵溶液(2×100 mL),饱和食盐水(2×100 mL)萃取,合并有机相,用无水硫酸钠干燥,减压蒸除溶剂,粗品经硅胶柱层析纯化得淡黄色固体2a,产率81.6%, Rf=0.38(石油醚/乙酸乙酯=3/1,V/V), m.p.73.9~75.0 ℃;1H NMR(500 MHz,CHLOROFORM-d)δ: 7.53(d,J=7.6 Hz, 2H), 7.43~7.49(m,J=7.6 Hz, 2H), 7.37(t,J=7.2 Hz, 2H), 7.17~7.24(m,J=7.3 Hz, 2H), 7.04~7.15(m, 2H), 3.71(t,J=6.1 Hz, 2H), 2.74(t,J=7.5 Hz, 2H), 1.93(quin,J=6.9 Hz, 2H), 1.62(br s, 1H), 1.37(br. s, 1H), 0.10(s, 1H);13C NMR(126 MHz)δ: 141.4, 137.5, 135.1, 128.8, 128.6, 128.5, 128.0, 127.4, 126.6, 126.4, 77.3, 76.7, 62.2, 34.1, 31.8, 1.0; IRν: 3263, 3023, 2938, 2863, 2363, 1594, 1447, 1058,1016, 967, 812, 749, 692, 529 cm-1; LC-MSm/z: calcd for C17H18O{[M+H]+}239.1436, found 239.1427。
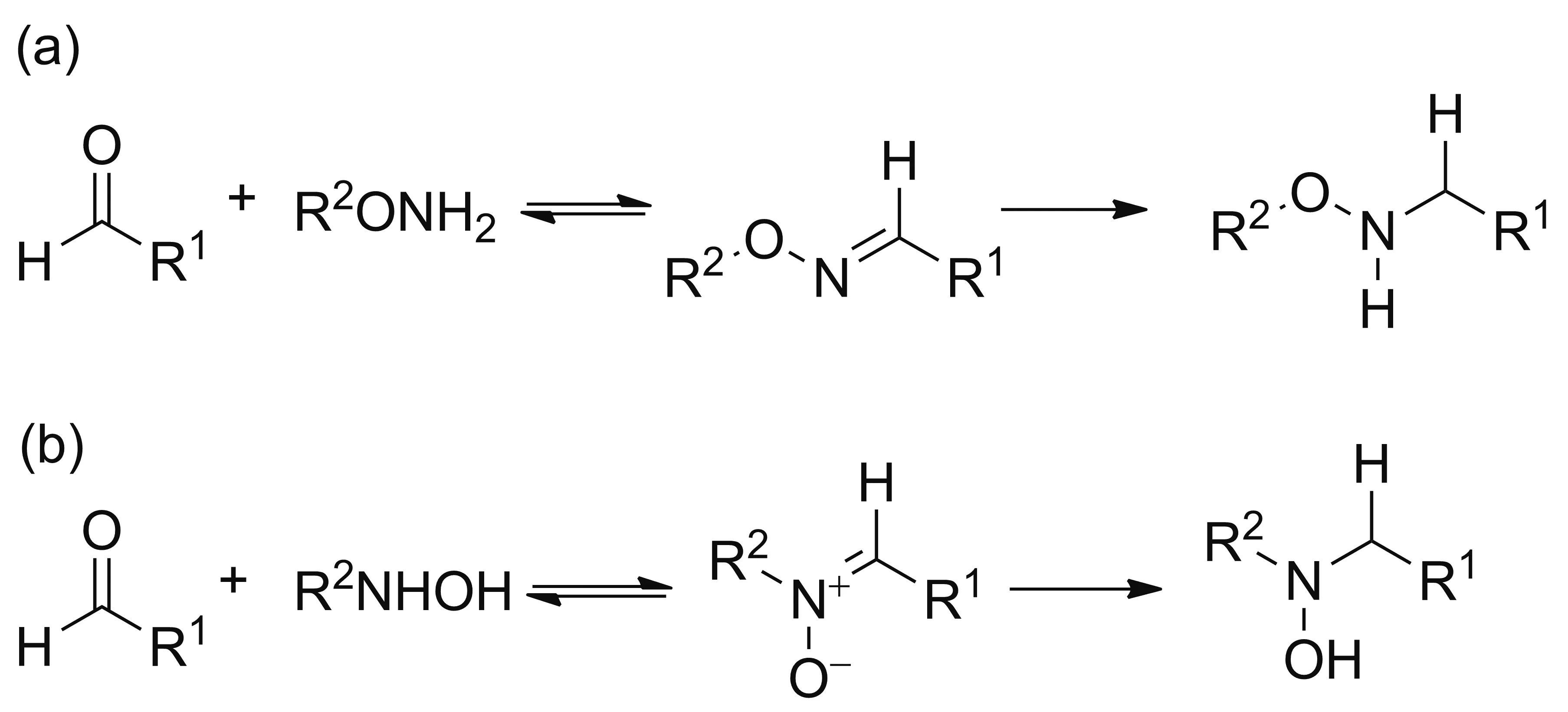
Chart 1
Scheme 1
(2)3a的合成
依次将化合物2a0.10 g(0.42 mmol)、 IBX 0.24 g(0.84 mmol)加入13 mL叔丁醇中,回流(82 ℃)反应4 h(TLC检测)。经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=6/1,V/V)纯化得淡黄色粉末3a,产率64.6%, Rf=0.5(石油醚/乙酸乙酯=5/1,V/V), m.p.55.8~58.0 ℃;1H NMR(500 MHz, DMSO-d6)δ: 9.72(s, 1 H), 7.58(d,J=7.3 Hz, 2H), 7.52(d,J=7.9 Hz, 2 H), 7.37(t,J=7.5 Hz, 3H), 7.16~7.32(m, 4H), 2.87(t,J=7.3 Hz, 2H), 2.78(t,J=7.3 Hz, 2H);13C NMR(126 MHz , DMSO-d6)δ: 203.2, 140.8, 137.6, 135.3, 129.1, 129.1, 128.7, 128.2, 128.0, 127.0, 126.8, 44.7, 40.5, 40.3, 40.2, 40.0, 39.8, 39.7, 27.7; IRν: 3024, 2929, 2850, 2820, 2721, 1713, 1595, 1513, 1489, 1449, 1405, 1388, 969, 819, 753, 691, 548 cm-1; LC-MSm/z: calcd for C17H16O{[M+H]+}237.1279, found 237.1273。
(3)4a的合成
将化合物3a100.00 mg(0.43 mmol)加入混合溶剂(四氢呋喃/水=1/1,V/V)4 mL中,加入盐酸羟胺45.00 mg(0.65 mmol),用2 mol·L-1NaOH溶液调至pH 4,于室温反应至终点(TLC检测)。旋蒸除溶,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=6/1,V/V)纯化得白色固体4a,产率45.7%, Rf=0.33(石油醚/乙酸乙酯=5/1,V/V), m.p.101.2~102.4 ℃;1H NMR(500 MHz, DMSO-d6)δ: 10.81(s, 1H), 7.59(d,J=7.3 Hz, 2H), 7.53(d,J=7.9 Hz, 2H), 7.37(t,J=7.6 Hz, 3H), 7.19~7.31(m, 4H), 6.68(t,J=5.2 Hz, 1H), 2.72~2.79(m, 2H), 2.53~2.60(m, 2H);13C NMR(126 MHz, DMSO-d6)δ: 150.0, 141.3, 137.6, 135.3, 129.2, 129.1, 128.7, 128.2, 128.0, 127.0, 126.9, 40.5, 40.3, 40.2, 40.0, 39.8, 39.7, 31.7, 26.6; IRν: 3209, 3087, 3027, 2871, 2362, 1665, 1490, 1447, 1324, 1073, 966, 691 cm-1; LC-MSm/z: calcd for C17H17NO{[M+H]+}252.1388, found 252.1379。
(4)5a和5b的合成(以5a为例)
将化合物4a33.7 mg(0.13 mmol)溶解于1 mL甲醇中,分批加入NaCNBH346.5 mg(0.74 mmol),用氯化氢的饱和甲醇溶液调至pH为弱酸性,反应6 h。旋蒸除溶,残余物预过柱,收集有机层再经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2/1,V/V)纯化得化合物5a,产率20%, Rf=0.3(石油醚/乙酸乙酯=1/1,V/V), m.p.111.2~113.0 ℃;1H NMR(500 MHz, DMSO-d6)δ: 7.58(d,J=7.3 Hz, 2H),7.41~7.54(m, 2H), 7.36(t,J=7.5 Hz, 2H), 7.09~7.31(m, 5H), 2.67~2.76(m, 1H), 2.57~2.65(m, 2H), 1.64~1.88(m, 3H);13C NMR(126 MHz, DMSO-d6)δ: 141.2, 141.0, 137.6, 135.4, 131.7, 131.1, 129.2, 129.2, 128.7, 128.2, 128.0, 128.0, 127.1, 127.0, 126.9, 126.8, 56.7, 56.6, 40.5, 40.3, 40.2, 40.0, 39.7, 32.3, 31.9, 26.5, 26.4; IRν: 3023, 2854, 2364, 2329, 1726, 1549, 1452, 1373, 1265, 1105, 1075, 964, 809 cm-1; LC-MSm/z: calcd for C17H19NO{[M+H]+}254.1545, found 254.1539。
用类似的方法合成5b, Rf=0.3(石油醚/乙酸乙酯=1/1,V/V), m.p.148.2~150.3 ℃;1H NMR(500 MHz, DMSO-d6)δ: 9.59, 9.17(d,J=4.0 Hz, 1H), 7.56~7.51(m,J=7.9 Hz, 2H), 7.24~7.19(m,J=8.2 Hz, 2H), 2.94~2.81(m, 2H), 2.68~2.57(m, 2H);13C NMR(126 MHz, DMSO-d6)δ: 140.6, 136.9, 134.8, 128.6, 56.2, 40.0, 39.8, 39.7, 39.3, 39.2, 39.0, 31.8, 25.9; LC-MSm/z: calcd ford7-C17H12NO{[M+H]+}261.1984, found 261.1982。
1.3 性能测试
(1) UV-Vis
分别使用乙腈和DMSO配制5.0 μmol·L-1化合物5a的溶液。并分别以乙腈和DMSO作为空白对照,在260~400 nm扫描UV-Vis谱图。
(2) FL
使用DMSO配制1.0 μmol·L-1化合物5a的溶液,在230~500 nm扫描FL谱图。
(3) 荧光量子产率[21]
配制1.0μmol·L-1化合物5a的DMSO溶液作为待测溶液,2.0 μmol·L-1硫酸奎宁的0.1 mol·L-1硫酸溶液作为标准溶液,测定吸光度和折射率,并计算发射峰积分面积。
(4)5a与麦芽三糖衍生体系的荧光性能
将20 μL 化合物5a(1 mmol·L-1)、 10 μL 0.01 mmol·L-1DP3溶液和10 μLNH4Ac/HAc(0.2 mol·L-1, pH=3.5)混合均匀,于60 ℃反应5 h。每隔1 h取样,在230~500 nm扫描FL谱图。
(5)5a与聚糖衍生反应
将20 μL 化合物5a(1 mmol·L-1)、 10 μL 0.01 mmol·L-1聚糖混合溶液(DP3、 DP5和DP7)与10 μL NH4Ac/HAc(0.2 mol·L-1, pH=3.5)混合均匀,于60 ℃反应3 h。浓缩,真空干燥,加入10%乙腈溶液,经0.22 μm尼龙滤膜过滤,进行质谱检测。
(6)1H和D标记的二苯乙烯羟胺化合物与DP5衍生反应
将20 μL同位素标记混合物(化合物5a/5b=1/1, 1 mmol·L-1)、 10 μL 0.01 mmol·L-1DP5溶液和10 μL NH4Ac/HAc(0.2 mol·L-1, pH=3.5)混合均匀,于60 ℃反应3 h。浓缩,真空干燥,加入10%乙腈溶液,经0.22 μm尼龙滤膜过滤,进行质谱检测。
2 结果与讨论
2.1 合成
以4-溴苯丙醇为初始原料,尝试以两种不同的Heck反应条件合成化合物2a。首先用醋酸钯,三苯基膦和N,N-二异丙基乙胺作催化剂,产率较低(23.8%);后来采用醋酸钯,四丁基溴化铵和碳酸铯作催化剂,产率较高(81.6%)。原料中的溴原子为Heck反应提供了便利条件,确保醋酸钯中的钯元素由二价态转化为零价态,使得催化反应可以顺利进行。虽然构建二苯乙烯结构乙烯桥的方法有很多,如Wittig反应[22],Heck反应[23]和Suzuki反应[24]等,但文献[25-26]表明使用Heck反应合成E-型苯乙烯基化合物的效率最高。
化合物3a可以通过氧化2a得到。本文采用氯铬酸吡啶盐(PCC)和邻碘酰基苯甲酸两种不同的氧化剂进行了氧化反应。以PCC为氧化剂时,反应时长为3 h,产率为40%;以邻碘酰基苯甲酸为氧化剂时,反应时长缩短至2 h,产率升高至64.6%,且操作简便。因此最终采用邻碘酰基苯甲酸作为氧化剂。
化合物3a经过肟化反应和还原反应分别得到化合物4a和5a。由于反应原料在氯化氢/甲醇饱和溶液中的溶解度不高,导致反应产率较低。由化合物5a的傅里叶红外光谱可知,其烯烃结构的=C—H面外摇摆振动在964 cm-1处有一个强的吸收峰,在730~650 cm-1没有弱且宽的吸收峰,因此可判定其双键为反式结构。
2.2 紫外-可见吸收光谱
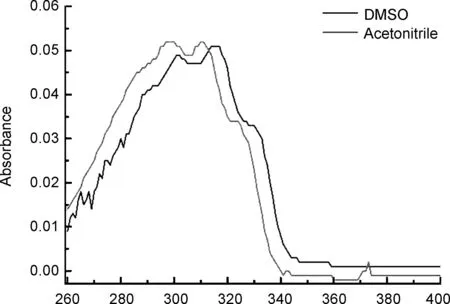
化合物5a的UV-Vis谱图如图1所示。由图1可见,化合物5a在乙腈和DMSO中的主要特征吸收峰均为两个,这证明了分子内存在不同类型的电子跃迁。化合物5a在DMSO中的吸收峰位于302 nm和312 nm,在乙腈中位于300 nm和310 nm。受溶剂效应影响,化合物5a在乙腈中的吸收峰相比于DMSO中的吸收峰发生了蓝(紫)移,吸收峰强度也更强。
λ/nm
2.3 荧光光谱和荧光量子产率
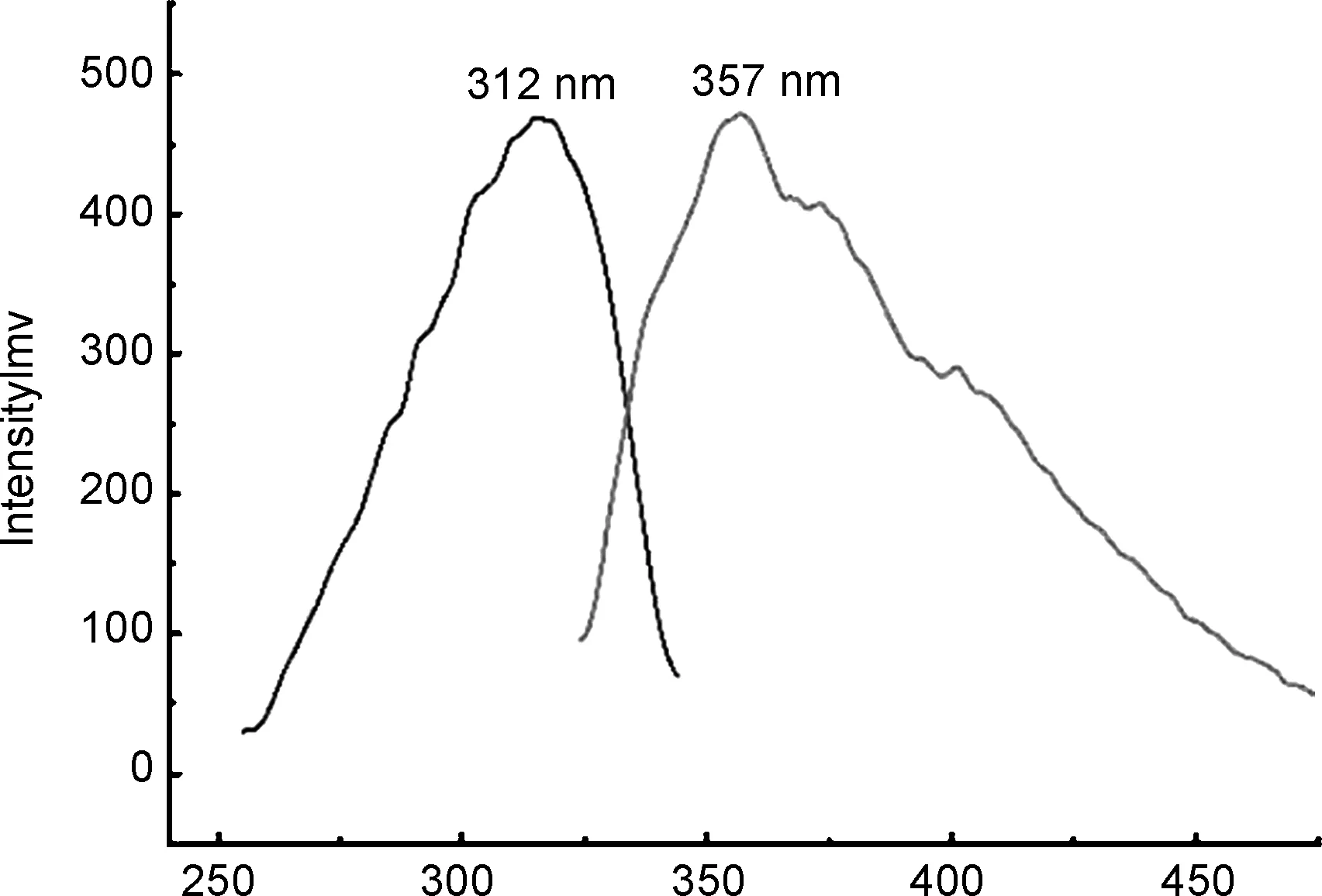
化合物5a在DMSO中的荧光激发和发射光谱如图2所示。其激发波长为312 nm,发射波长为357 nm,斯托克斯位移为45 nm(表1)。使用硫酸奎宁作为标准品,测得化合物5a的荧光量子产率为0.11,摩尔吸光系数为1.02×104cm-1·mol-1·L。
λ/nm

表1 化合物4的物理化学性质
2.4 5a与麦芽三糖衍生体系荧光随时间的变化
化合物5a荧光性能较好,斯托克斯位移为45 nm,且具有较强的抗背景干扰能力。我们期望化合物5a与糖的衍生产物也具有良好的荧光性能。但是由图3可以看出,随着反应时间的延长,反应体系的荧光强度在逐渐降低,3 h之后荧光强度几乎不再改变,说明化合物5a与麦芽糖的衍生反应没有生成荧光性能显著增强的衍生产物。
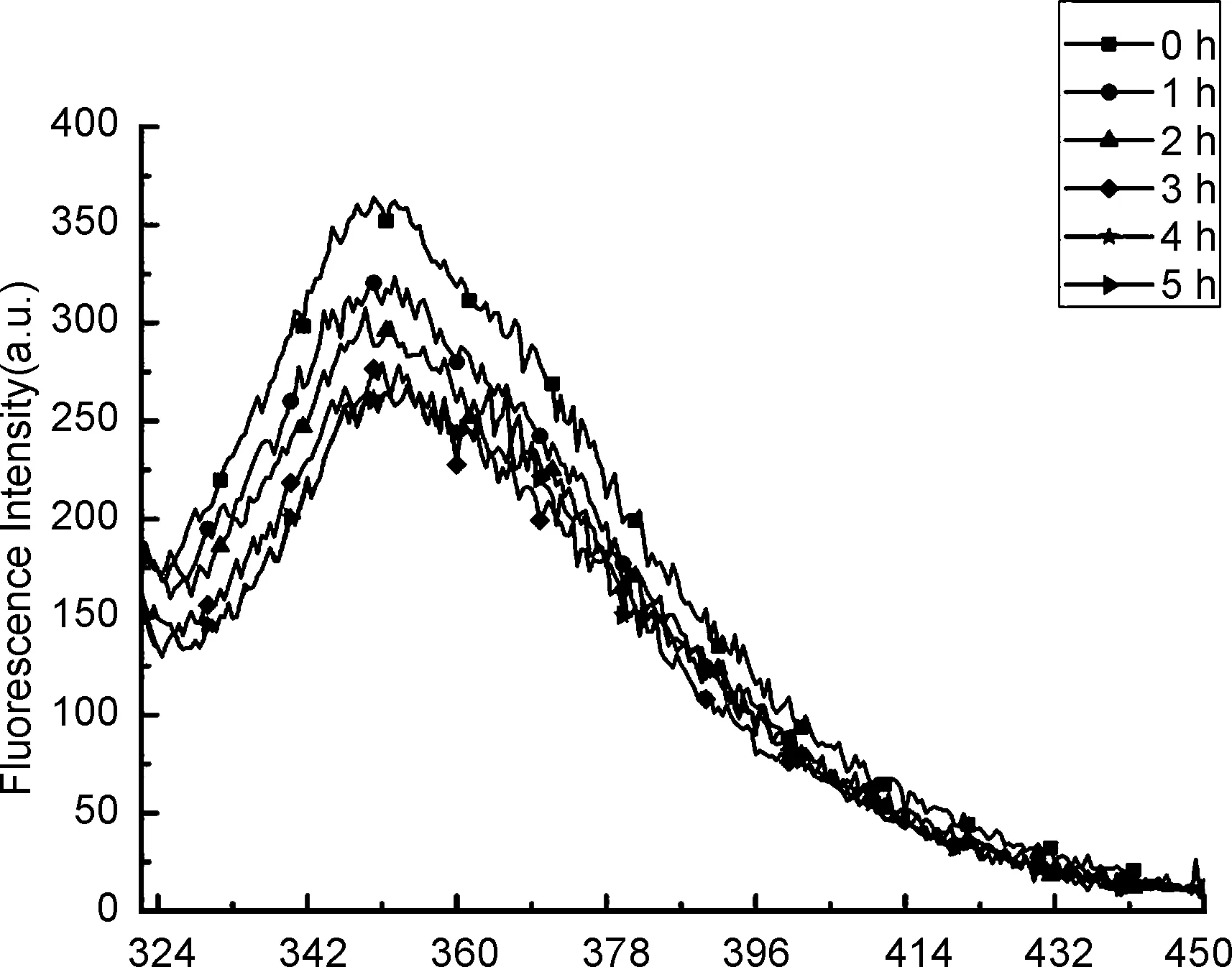
λ/nm图3 5a与DP3衍生体系的FL谱图
2.5 聚糖衍生物的质谱分析
虽然化合物5a很难用于聚糖分子基于荧光衍生的相关研究,但仍能通过衍生产物的质谱来实现聚糖的检测和定量分析。化合物5a分别与麦芽三糖(DP3)、麦芽五糖(DP5)和麦芽七糖(DP7)进行衍生化反应后,所得到的衍生产物对应的质谱峰强度较高(图4)。质谱结果显示,化合物5a与聚糖衍生同时被还原,衍生产物质谱峰的质荷比相较于理论值增加了两个氢原子的原子量(表2),这可能是衍生反应得到的硝酮在质谱离子化过程中进一步还原的结果(图5)。
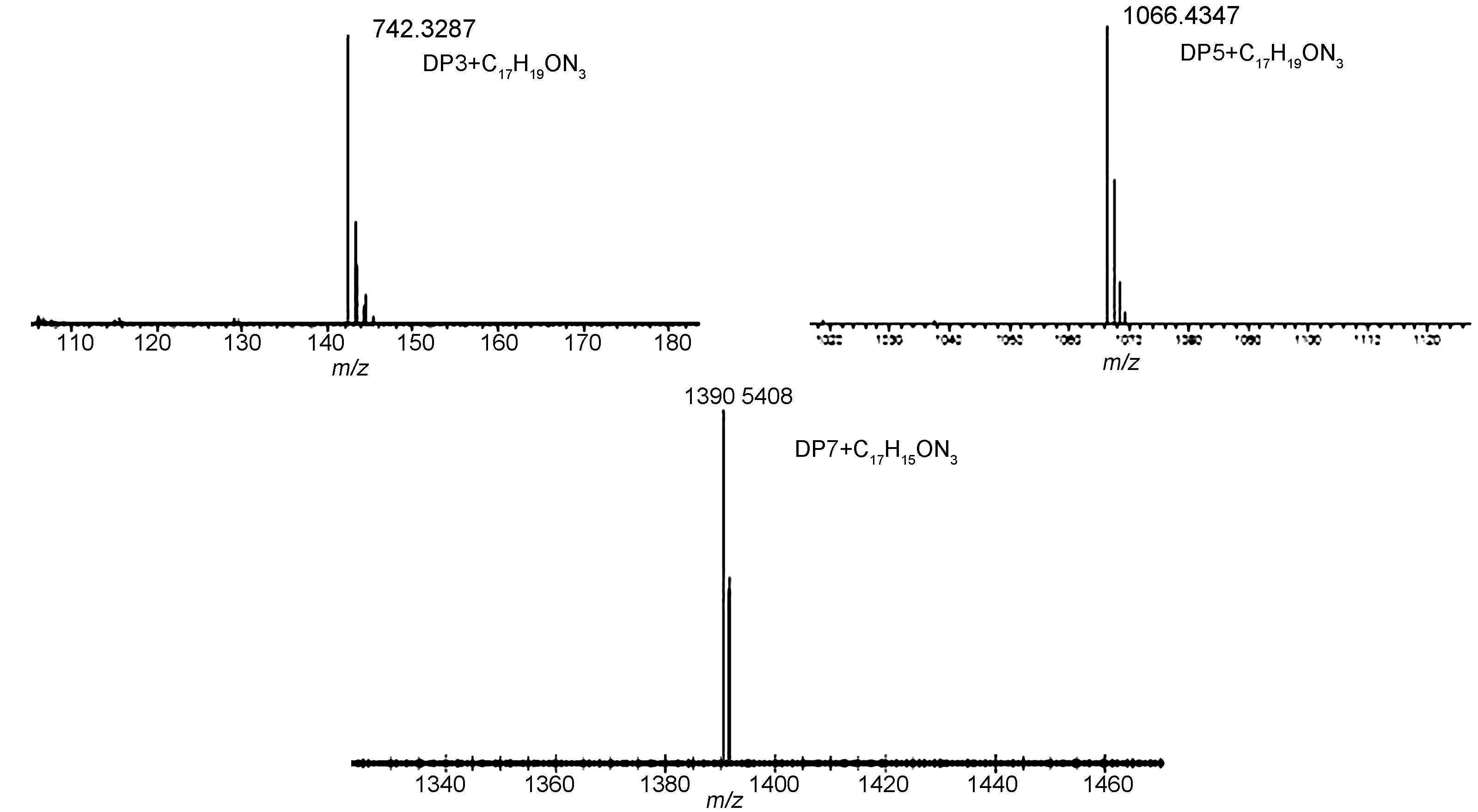
图 4 化合物5a与DP3、 DP5、 DP7衍生物的MS谱图
Figure 4 MS spectrometry of the derivatives of 5a and DP3, DP5, DP7
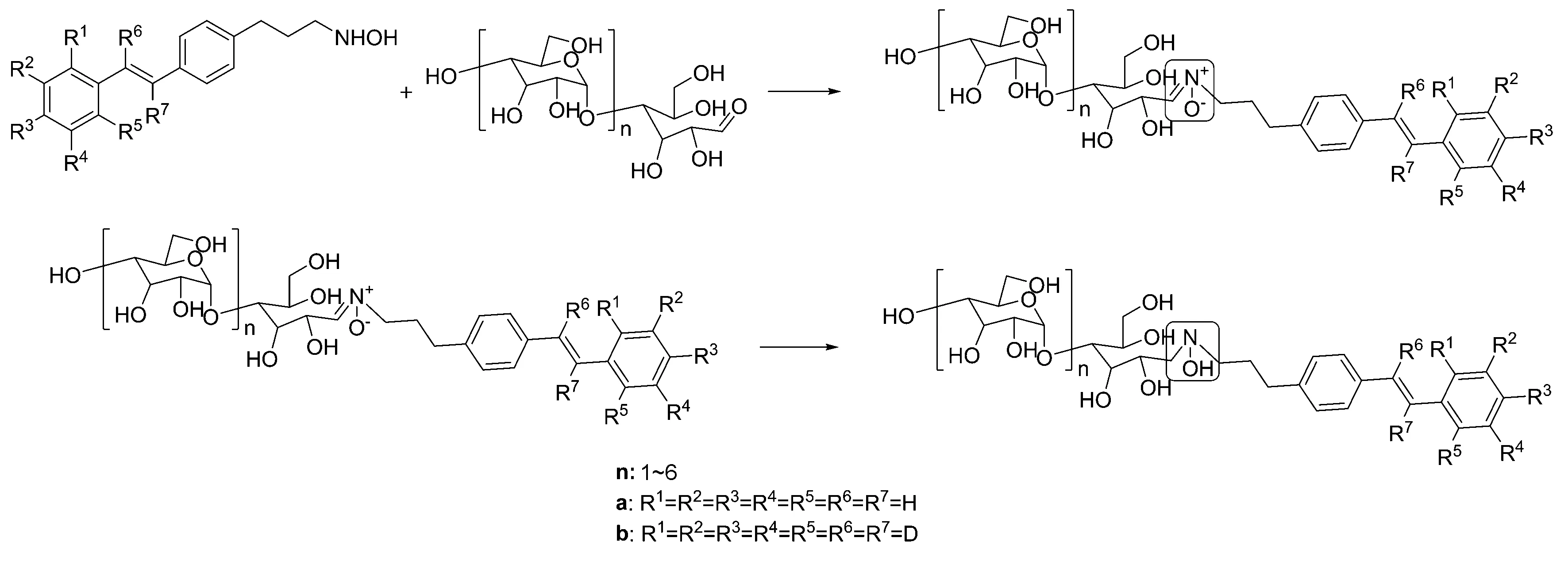
图5 化合物5a与聚糖的硝酮化反应和还原反应
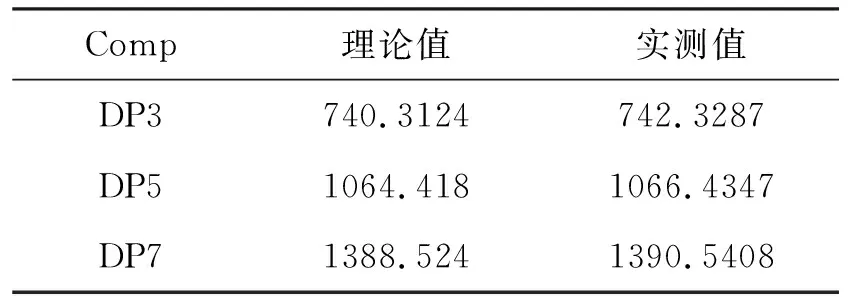
表 2 化合物5a与聚糖衍生物的质谱数据
2.6 1H和D标记的二苯乙烯羟胺化合物与DP5衍生反应的质谱
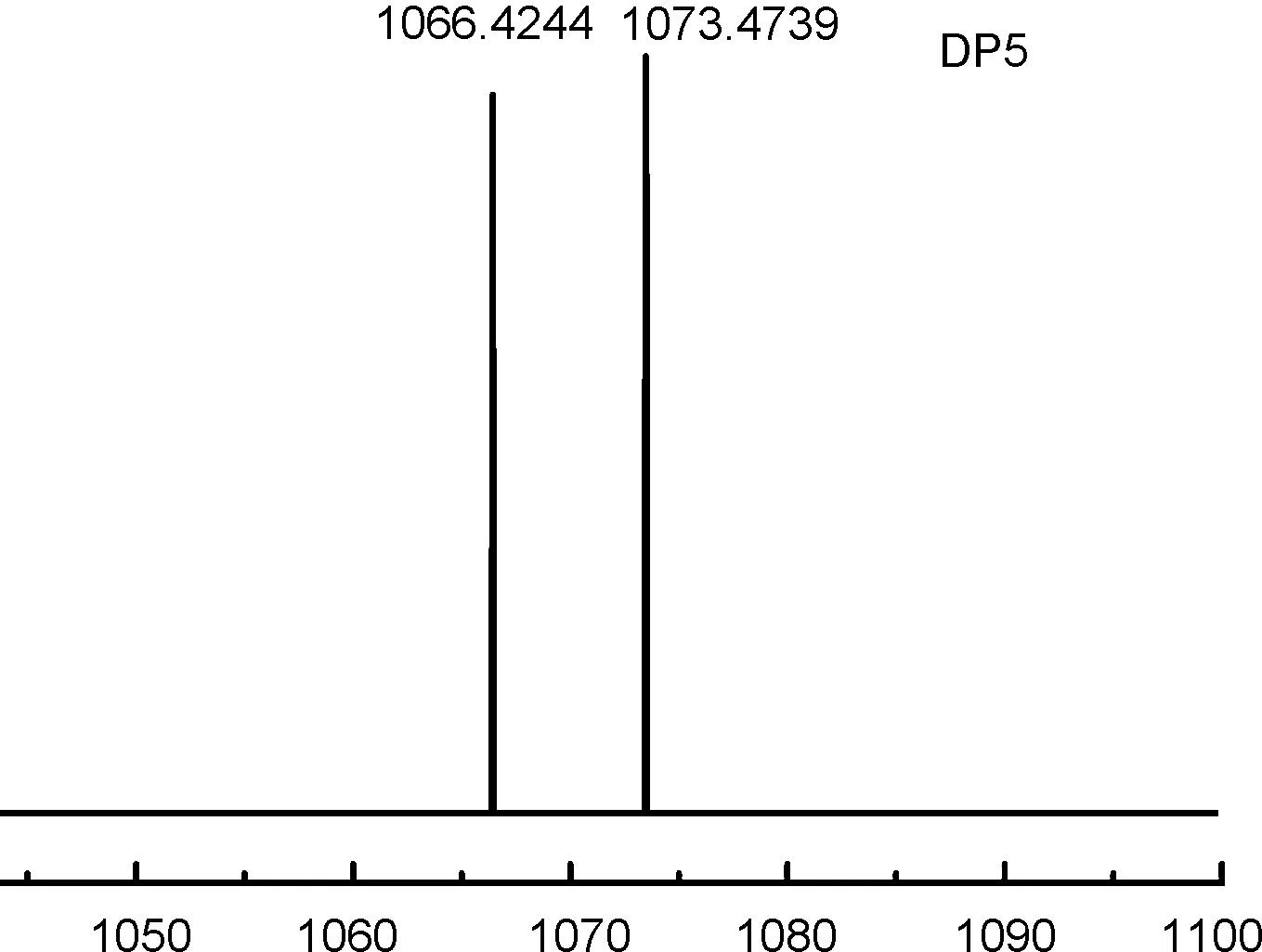
近年来,稳定同位素标记的试剂已被证明是对简单和复杂混合物中N-连接聚糖进行相对定量的可行策略[21-24]。化合物5a和5b等量混合后与DP5进行衍生化反应,得到的衍生物的质谱图如图6所示。5a和5b与DP5的衍生物加氢峰的质荷比分别为1066.4244和1073.4739,相差为7,强度比接近1。衍生条件相对温和,质谱信号的强度高,“轻”质和“重”质同位素标记的二苯乙烯羟胺探针能够较好的标记聚糖,衍生产物的离子化效果基本相同,可以将成对出现、质荷比相差为7的质谱峰的信号强度用于定量分析的依据。这说明该类探针在聚糖的定量分析中有比较广阔的应用前景。
m/z
设计并合成了新型二苯乙烯类羟胺化合物(化合物5a,5b)。化合物5a的激发波长为312 nm,发射波长为357 nm,斯托克斯位移为45 nm,荧光量子产率为0.11,在发生衍生反应之前具有较好的荧光性能。5a和5b按1/1与麦芽五糖进行衍生化反应时,得到了成对出现,分子量相差为7的稳定同位素标记产物。这些质谱信号的强度可以作为定量分析的依据,“轻”质和“重”质同位素标记的化合物5a和5b联合使用时,可以基于硝酮化反应和质谱技术,在聚糖和其它醛类分子的定量分析中发挥作用。我们将在后期的工作中引入四苯乙烯或其它荧光性能更好的砌块,从而得到既能作为荧光探针,又可以用于质谱分析的多功能分子,进一步拓展其在糖类检测中的应用。
