益肾化湿颗粒质量标准提高研究
2020-07-15卢秋梅黄艳霞栗建明
卢秋梅,黎 倩,黄艳霞,栗建明
(1. 广州市药品检验所·国家药品监督管理局中成药质量评价重点实验室,广东 广州 510160;2. 广州康臣药业有限公司,广东 广州 510635)
益肾化湿颗粒为广州康臣药业有限公司独家品种,处方源自《脾胃论》中的升阳益胃汤,由人参、黄芪、黄连等16 味中药组方[1],功效为升阳补脾、益肾化湿、利水消肿,用于脾虚湿盛证所致蛋白尿,兼见水肿、疲倦乏力、畏寒肢冷、纳少等证[2]。该方传统剂型为汤剂,不易贮存及携带,颗粒剂克服了汤剂的不足,具有稳定、易携带的特点[3]。目前,该药仅有局颁标准(国家食品药品监督管理局标准YBZ04482009)。在此基础上,本研究中新增了人参的薄层色谱(TLC)鉴别和黄芪中黄芪甲苷的高效液相色谱(HPLC)法含量测定,修订了人参的HPLC 含量测定方法(增加人参皂苷Rg1和Re 指标),完善了黄芪、独活、甘草、白芍、黄连和陈皮的TLC 鉴别方法。现报道如下。
1 仪器与试药
1.1 仪器
P300H 型超声波清洗器(德国Elma 公司,功率为300 W,频率为37 kHz);TLC 板加热器,TLC Visualizer薄层成像系统(瑞士Camag 公司);XP26 型电子天平(精度为0.1 mg);MS204S 型电子天平(精度为0.001 mg),均购自瑞士Mettler Toledo 公司;ST16 型高速离心机(美国赛默飞公司);TW12 型恒温水浴锅(德国Julabo 公司);硅胶G60 薄层板(德国Merck 公司);1260Ⅱ型高效液相色谱仪(美国安捷伦公司),包括Alltech 6000 蒸发光散射检测器,Openlab CDS 2.2 色谱工作站。
1.2 试药
益肾化湿颗粒(批号分别为20180608,20180609,20180701,规格为每袋10 g)及所有阴性样品均购自广州康臣药业有限公司;对照药材黄芪(批号为120974 -201612)、人参(批号为120917 -201712)、独活(批号为120940 -201612)、甘草(批号为120904 -201620)、白芍(批号为120905 -201610)、黄连(批号为120913 -201611)、陈皮(批号为120969 -201510),以及对照品人参皂苷Rg1(批号为110703 -201832)、人参皂苷Re(批号为110754 -201827)、人参皂苷Rb1(批号分别为110704 -201726,110704 -201827)、黄芪甲苷(批号为110781 -201717)、芍药苷(批号为110736 -201842)、盐酸小檗碱(批号为110713 -201814)、橙皮苷(批号为110721 -201818)、二氢欧山芹醇当归酸酯(批号为111583 -201304),均购自中国食品药品检定研究院;乙腈为色谱纯,其他试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 TLC 鉴别
人参、黄芪:取本品4 g,加水40 mL 溶解,离心,取上清液,用乙醚萃取3 次,每次30 mL,合并乙醚液(备用),分取水溶液(溶液1)10 mL,用水饱和正丁醇振摇提取3 次,每次10 mL,合并正丁醇液,用氨试液提取3 次,每次30 mL,弃去氨试液,正丁醇蒸干,残渣加甲醇2 mL 使溶解,作为供试品溶液。取人参对照药材0.5 g,黄芪对照药材1 g,同法制成对照药材溶液。分别取缺人参、黄芪的阴性样品适量,同法制得缺人参、黄芪阴性对照品溶液。取人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1及黄芪甲苷对照品,加甲醇制成每1 mL 含1 mg 的混合溶液,作为对照品溶液。吸取上述溶液1 ~2 μL,分别点于同一硅胶G 薄层板上,以正丁醇-乙酸乙酯-稀氨液(5→50,4 ∶1 ∶5,V/ V/ V)的上层溶液为展开剂,另槽加入10 mL 浓氨试液,预平衡15 min,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰,置日光及紫外光(365 nm)下检视。色谱图见图1。可见,供试品溶液色谱中,在与对照药材溶液和对照品溶液色谱相应位置上分别显相同颜色的斑点或荧光斑点,阴性对照无干扰。

图1 人参、黄芪薄层色谱图
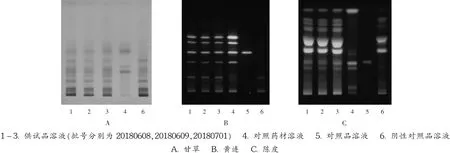
图2 甘草、黄连、陈皮薄层色谱图
甘草:取水溶液1,用水饱和正丁醇振摇提取3 次,每次20 mL,合并正丁醇液,正丁醇蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。取甘草对照药材0.5 g,加水40 mL,煎煮30 min,滤过,取滤液同法制成对照药材溶液。取缺甘草阴性样品适量,同法制得缺甘草阴性对照品溶液。吸取上述3 种溶液各5 μL,分别点于同一硅胶G 薄层板上使成条带状,以乙酸乙酯-冰醋酸-甲酸-水(15 ∶1 ∶1 ∶2,V/ V/ V/ V)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇,105 ℃加热至斑点显色清晰,日光下检视。色谱图见图2 A。可见,供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰。
黄连:取本品4 g,加95%乙醇15 mL,超声30 min,过滤,滤液浓缩至5 mL,作为供试品溶液(溶液2)。取黄连对照药材0.2 g,同法制成对照药材溶液。取缺黄连阴性样品适量,同法制得缺黄连阴性对照品溶液。取盐酸小檗碱对照品,加甲醇制成每1 mL 含0.5 mg 的溶液,作为对照品溶液。吸取供试品溶液、对照药材溶液及阴性样品溶液各4 μL、对照品溶液2 μL,分别点于同一硅胶G 薄层板上使呈条带状,以甲苯-乙酸乙酯-甲醇-异丙醇-水(6 ∶3 ∶2 ∶1.5 ∶0.3,V/ V/ V/ V/ V)为展开剂,另槽加入10 mL 浓氨试液,预平衡15 min,展开,取出,晾干,置紫外光(365 nm)下检视。色谱图见图2 B。可见,供试品溶液色谱中,在与对照药材溶液和对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照无干扰。
陈皮:取陈皮对照药材0.3 g,加95%乙醇15 mL,超声30 min,过滤,滤液蒸干,定容至5 mL,作为对照药材溶液。取缺陈皮阴性样品适量,同法制得缺陈皮阴性对照品溶液。取橙皮苷对照品,加甲醇制成每1 mL 含0.4 mg 的溶液,作为对照品溶液。吸取溶液2、对照药材溶液及缺陈皮阴性对照品溶液各4 μL、对照品溶液2 μL,分别点于同一硅胶G 薄层板上(条状点样),以三氯甲烷-甲醇-水(28 ∶10 ∶1,V/ V/ V)为展开剂,展开,取出,晾干。喷以5%三氯化铝乙醇溶液,在105 ℃加热数分钟,置紫外光(365 nm)下检视。色谱图见图2 C。可见,供试品溶液色谱中,在与对照药材溶液和对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照无干扰。
2.2 含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:Waters X select HSS T3 十八烷基硅烷键合硅胶柱(250 mm ×4.6 mm,5 μm);流动相:乙腈(A)-水(B),梯度洗脱程序见表1;蒸发光散射检测器,漂移管温度:105 ℃,载气流速:3.0 L/min。在此色谱条件下,各成分分离度良好,阴性对照无干扰,理论板数按人参皂苷Rg1峰计算应不低于6 000。色谱图见图3。
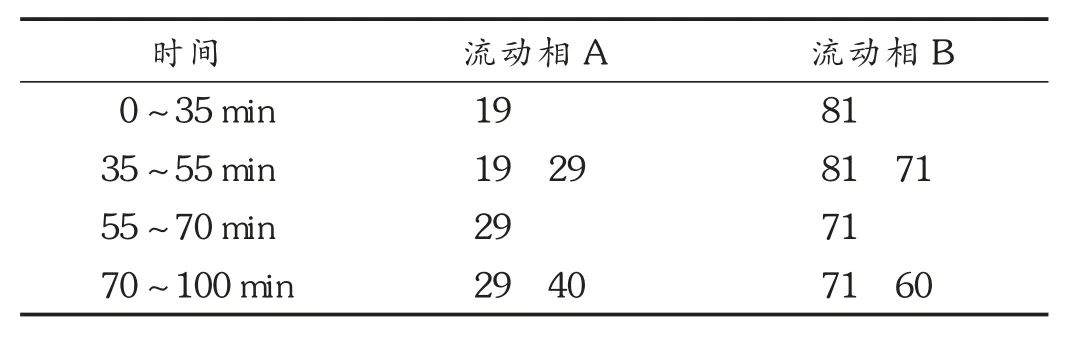
表1 流动相梯度洗脱程序(%)
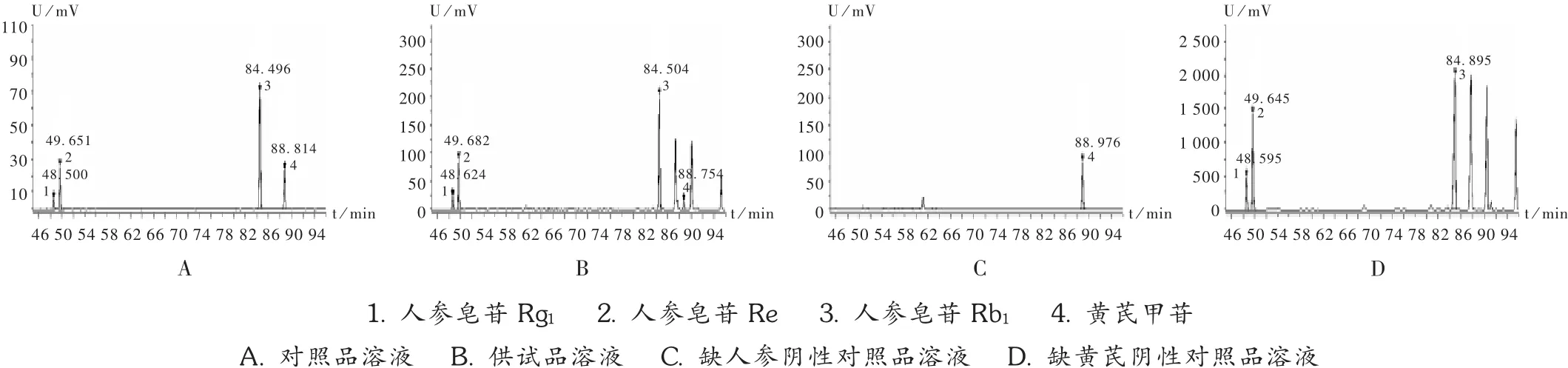
图5 含量测定高效液相色谱图
2.2.2 溶液制备
取各对照品适量,精密称定,加甲醇制成每1 mL 含人参皂苷Rg10.08 mg、人参皂苷Re 0.13 mg、人参皂苷Rb10.20 mg 和黄芪甲苷0.07 mg 的混合溶液,即得对照品溶液。取本品约4 g,混匀,研细,精密称定,置具塞锥形瓶中,精密加水50 mL,超声处理(功率为300 W,频率为37 kHz)30 min,放冷,再称定质量,用水补足减失的质量,离心10 min,取上清液25 mL,加乙醚振摇提取3 次,每次30 mL,弃去乙醚液,水液再用水饱和正丁醇提取5 次,每次25 mL,合并正丁醇提取液,用氨试液充分洗涤2 次,每次40 mL,合并氨试液,用水饱和正丁醇提取2 次,每次20 mL,合并正丁醇液,蒸干,残渣加甲醇溶解,转移至5 mL 容量瓶中,加甲醇至刻度,摇匀,即得供试品溶液。分别取缺人参阴性样品、缺黄芪阴性样品,按供试品溶液制备方法制备缺人参和缺黄芪阴性对照品溶液。
2.2.3 方法学考察
线性关系考察:配制线性关系考察样品溶液6 份,进样10 μL,以质量浓度的对数(X,mg/mL)为横坐标、峰面积的对数(Y)为纵坐标绘制校正曲线。结果见表2。
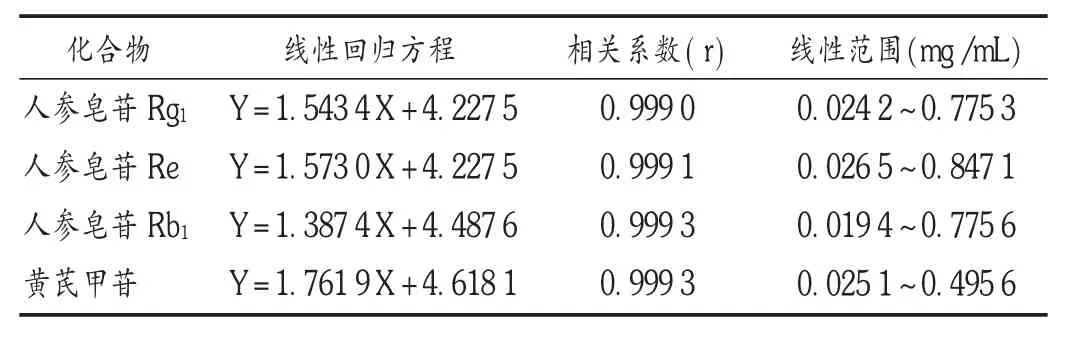
表2 各成分线性关系考察结果
重复性试验:取样品(批号为180608)6 份,精密称定,依法制备供试品溶液,按拟订色谱条件测定。结果人参皂苷Rg1,Re,Rb1及黄芪甲苷含量的RSD 分别为1.75%,3.33%,2.88%,3.07%(n =6),表明方法重复性良好。
稳定性试验:取样品(批号为180608)4 g,精密称定,依法制备供试品溶液,分别于0,4,8,12,16,20,24,28 h 时按拟订色谱条件测定。结果人参皂苷Rg1,Re,Rb1及黄芪甲苷含量的RSD 分别为3.71% ,2.40% ,1.81%,1.73%(n =8),表明供试品溶液28 h 内稳定。
加样回收试验:取样品(批号为180608)6 份,每份2 g,精密称定,置具塞锥形瓶中,精密加入混合对照品溶液(质量浓度分别为0.414 55,1.007 46,1.504 21,0.538 47 mg/mL 的人参皂苷Rg1,Re,Rb1和黄芪甲苷)1 mL,挥干,依法制备供试品溶液,按拟订色谱条件进样测定,计算回收率。结果见表3。
2.2.4 样品含量测定
为确保含量数据更具有代表性,广州康臣药业有限公司增加样品15 批,共18 批样品,依法制备供试品溶液,按拟订色谱条件进样测定,计算含量。结果见表4。按人参皂苷总量、黄芪甲苷含量平均值的80%设限,结果分别为1.08,0.26 mg /g。拟规定样品中每1 g 含人参以人参皂苷Rg1(C42H72O14)、人参皂苷Re((C54H92O23)和人参皂苷Rb1(C54H92O23)的总量计,不得少于1.08 mg;每1g 含黄芪以黄芪甲苷(C41H68O14)计,不得少于0.26mg。
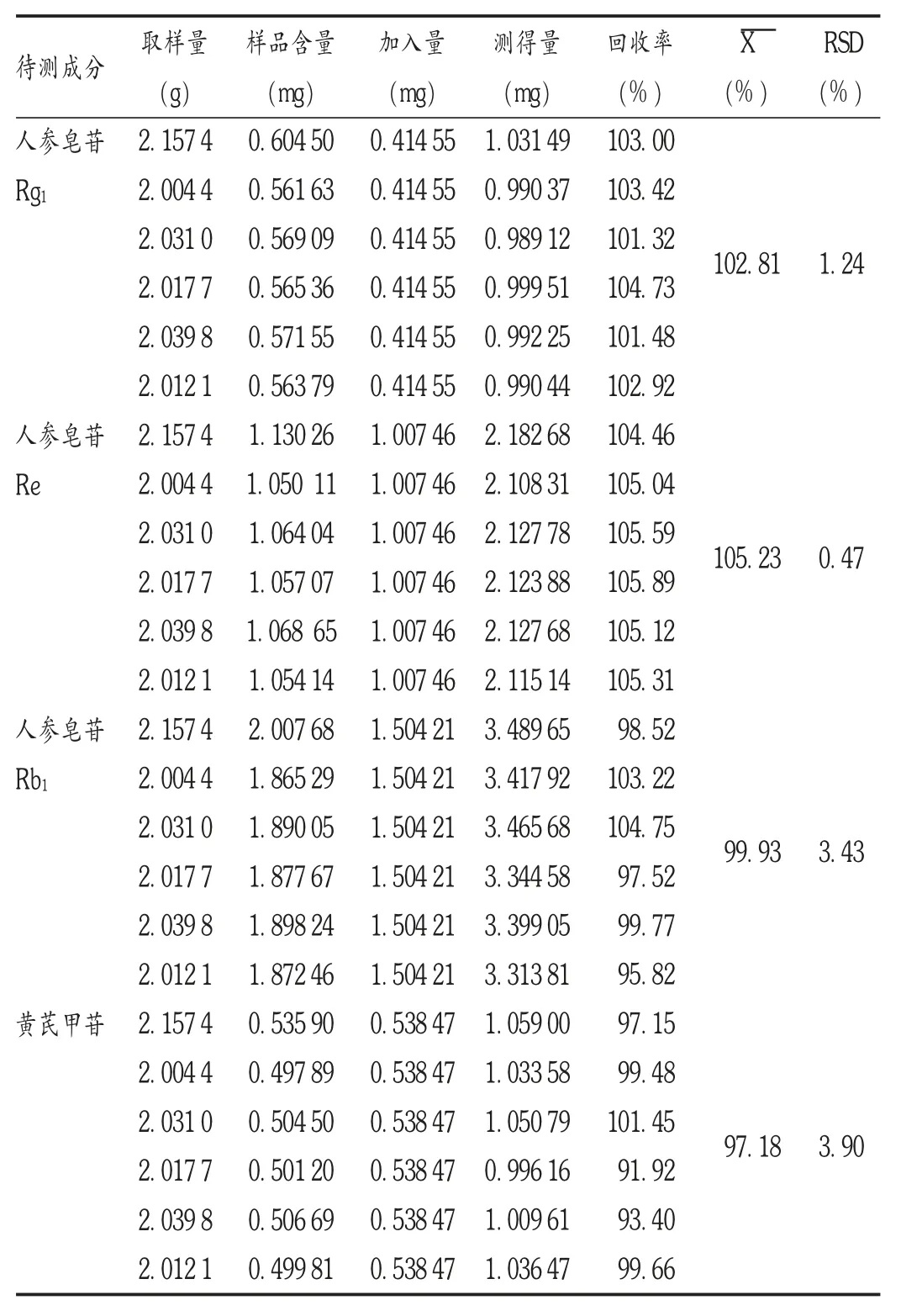
表3 各成分加样回收试验结果(n =6)
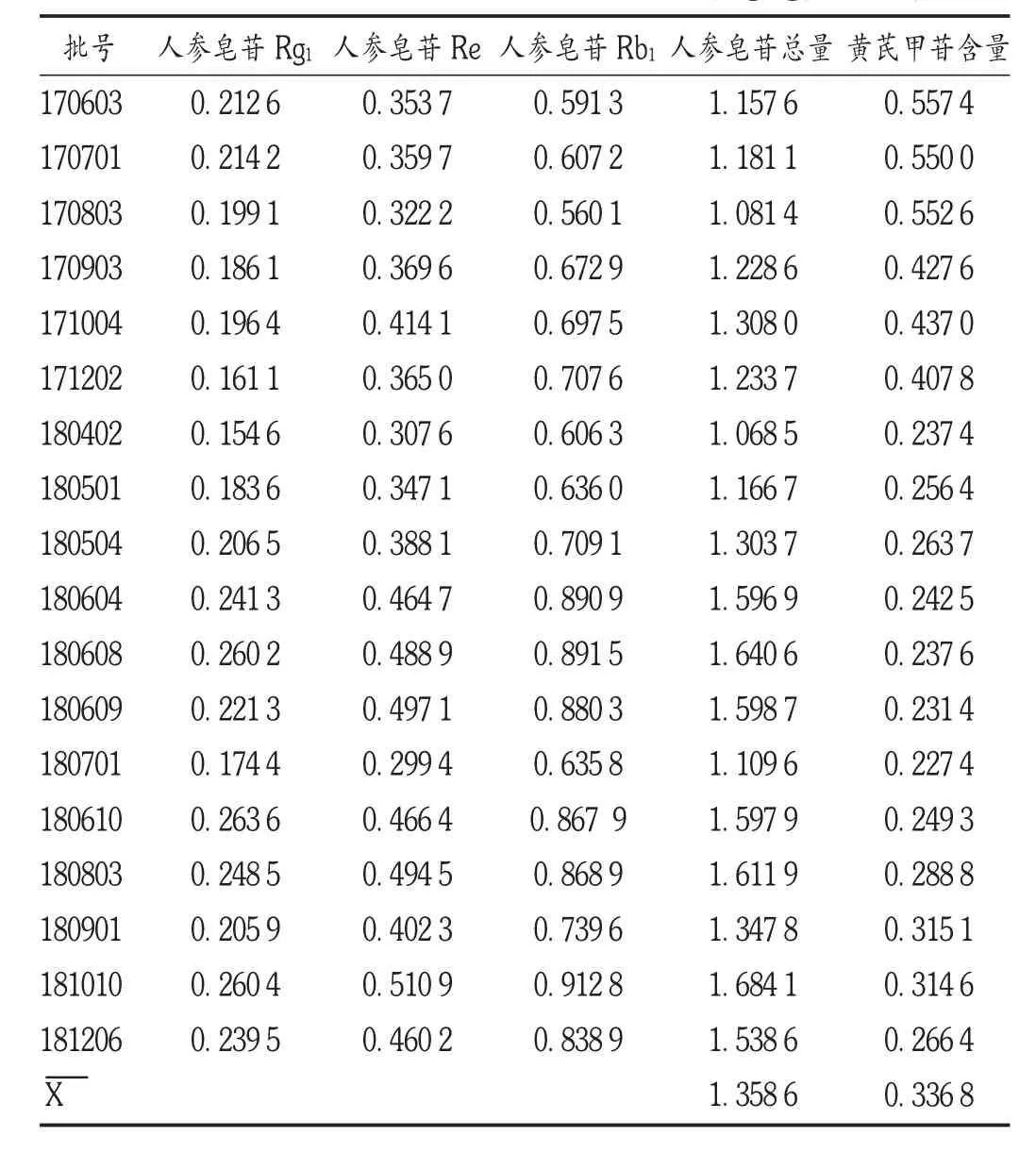
表4 18 批样品各成分含量测定结果(mg /g,n =18)
3 讨论
3.1 TLC 条件筛选
人参、黄芪:人参为新增项,黄芪为修订项。原标准黄芪鉴别采用萃取、洗涤后还需上大孔树脂柱多溶剂洗脱,操作烦琐,检验周期太长,故对原标准进行优化。人参、黄芪、三七等的皂苷鉴别常使用三氯甲烷-甲醇-水(13 ∶7 ∶2,V/ V/ V)及三氯甲烷-乙酸乙酯-甲醇-水(15 ∶40 ∶22 ∶10,V/ V/ V/ V)展开,难以将人参皂苷与黄芪甲苷较好地分离,且色谱条件受环境影响较大,需严格控制温度和湿度,重复性相对较差[4]。根据相关经验及参考文献[5],选择展开条件。
甘草:原标准以甘草酸铵对照品为对照,甘草阴性对照无干扰,但斑点信息较少,试验中曾喷以10%硫酸乙醇加热显色后再观察[6],发现甘草阴性对照有干扰,故参考文献[7]制订展开条件。
黄连:原标准中需要二次展开,操作较复杂,且加入黄连对照药材后,发现阴性对照有干扰,故参考文献[7]选择展开条件。
陈皮:修订时增加了陈皮对照药材,但在验证原标准方法时发现阴性对照有干扰。参考2015 年版《中国药典(一部)》女金丸项下的展开条件[8]521-522,修订后的方法操作简便,陈皮阴性对照无干扰,专属性强。
独活、白芍 独活:TLC 原标准的提取方式耗时长,且斑点不清晰,常需加大点样量,而采用2.1 项下乙醚液挥干后作为供试品溶液,既节省溶剂,减少样品用量,且斑点又清晰,阴性对照无干扰,方法专属性强。白芍在原标准基础上增加了白芍对照药材,阴性对照无干扰,方法专属性强。
3.2 TLC 条件考察
各TLC 鉴别均考察了青岛海洋化工硅胶G 和Merck 硅胶G60 预制板2 种不同薄层板的层析效果,各成分的Rf值存在一定差异,但与相邻成分分离度均良好,表明2 种薄层板均能满足鉴别的要求。另固定薄层板,比较在不同温湿度条件下展开的薄层效果,结果表明,温度和相对湿度对薄层色谱效果无明显影响。
3.3 HPLC 成分及色谱条件筛选
人参的主要有效成分为总皂苷,5 种人参皂苷(Rb1,Rb2,Rg1,Rc,Re)占总皂苷含量的80%,具有延缓疲劳产生的作用,主要表现在调节中枢神经系统、改善机体功能、消除疲劳等方面[9]。黄芪的主要有效成分为黄芪甲苷,黄芪甲苷治疗肾性高血压大鼠时,不仅降压效果显著,还可抑制肾氧化应激反应,缓解肾病理损伤[10]。人参、黄芪是益肾化湿颗粒的君药,其现代药理作用均与益肾化湿颗粒的功能主治密切相关。因此,对人参皂苷Rg1,Re,Rb1和黄芪甲苷进行定量及定性分析,可更好地控制该制剂的质量。
2015 年版《中国药典(一部)》中人参含量测定项下人参皂苷的测定波长为203 nm[8]8-9;黄芪含量测定项下黄芪甲苷含量测定采用蒸发光散射检测器法检测[8]302-303。最初采用紫外光与蒸发光串联同步测定,在研究过程中,发现样品在203 nm 波长处人参分离度较差、阴性对照有干扰,且人参皂苷Rg1,Re,Rb1在蒸发光下响应值也较高,而原标准中在测定人参中的人参皂苷Rb1时也采用蒸发光散射检测器法,最终确定文中的色谱条件。
