基于分子模拟技术研究来源于嗜热地衣芽孢杆菌葡聚糖酶的活性位点
2020-07-13禹亚男
禹亚男,李 婷
(重庆医科大学附属第二医院 医学重症监护科,重庆 400000)
嗜热酶是从嗜热微生物中分离得到的一类热稳定性酶,具有化学催化剂无法比拟的优点,尤其是在高温条件下保持的极好稳定性,克服了常温酶在应用中出现的化学性质不稳定的现象,使很多高温化学反应得以实现,从而将极大的促进生物技术产业的发展。葡聚糖酶(glucanase)能专一性催化葡聚糖或地衣多糖中临近β-1,3键、β-1,4键水解,产生3- 5个葡萄糖单位的低聚糖及葡萄糖,从而降低葡聚糖的亲水性和粘性[1],因此现被很多国家用来作为饲料添加剂[2]及啤酒生产的酶制剂[3]。对于来自不同物种的糖苷水解,其葡聚糖酶的氨基酸组成和的活性部位存在差异。例如,姜彤等研究来源于柄孢霉的β-葡聚糖酶晶体属于GH131糖苷酶家族,由外围的9个β折叠和内部的6个β折叠构成具有典型的β-jelly-roll结构,表面呈凹槽结构,易于和底物结合。其中E99和E139作为酸碱供体,是保守的催化结合位点[4]。来源于嗜热拟青霉的Ptlic16A晶体属于糖苷水解酶GH16家族,其催化结构为β-jelly-roll,其中E113作为亲核剂,E118作为质子供体,W97、W101、W253和底物的识别有关[5]。来源于黄孢原毛平革菌的Lam55A葡聚糖酶属于GH55家族,其具有两个平行的右手β折叠结构域构成的一个类似于胸腔的结构,其催化位点位于两个结构域之间[6]。植物葡聚糖酶主要属于GH17家族,其结构具有典型的TIM桶结构(TIM-barrel fold),其酸碱催化、亲核体分别位于第4和第7 β折叠链上[7]。来源于GH64家族的LPHase葡聚糖酶晶体其催化部位由一个β折叠和一个α/β结构域构成,在两个结构域之间形成了宽广的沟槽[8]。目前尚没有关于来源嗜热地衣芽孢杆菌葡聚糖酶晶体结构的报道。有研究表明,葡聚糖酶的最适温度为55℃,在温度100℃下,30 min仍有70%的生物活性并且可以催化多种糖苷键水解[9],是一种新型的葡聚糖酶。本研究在此研究的基础之上,通过利用分子对接与分子动力学模拟技术研究葡聚糖酶与二聚糖UNK之间分子的识别机制,以期为嗜热地衣芽孢杆菌葡聚糖酶分子结构后续的理性改造和高效利用提供有力依据。
1 材料与方法
1.1 葡聚糖酶结构的制备
利用在线服务器I-TASSER(http://zhanglab.ccmb.med.umich.edu/I-TASSER/)对嗜热地衣芽孢杆菌葡聚糖酶(登录号:Y_006712752.1)进行同源建模获的试验模板,通过GROMACS4.6.5软件[10]对其进行结构优化,获得最佳的构象。
1.2 配体分子的制备
利用ChemOffice 3D软件[11]构建二聚糖分子结构,用ChemOffice 3D的MM2模板对底物进行能量最小化,保存结构为pdbqt格式,以便进行进一步的对接工作。
1.3 分子对接
采用AutoDock4.2程序包[12]对二聚糖和葡聚糖酶进行预处理。对接时,在格点盒子中,X维、y维、z维格点数目分别设为52、54、54.该格点盒子的中心坐标为-24.855,14.414,93.059,格点间距为0.0392 nm。采用拉马克遗传算法(LGA)来寻找配体和受体最佳的结合位置,将局部能量搜索与遗传算法相结合,以半经验势函数为打分函数,对小分子构象和位置进行全局搜索。为了使结果更加准确,采用遗传算法中较大的运算参数,种群个数设为150,能量评估的最大数目设为1×107,运算循环数设为1000,其他的使用该程序默认的参数。同时设立2组平行组。最后依据获得的1000个配体小分子构象进行聚类分析(参数为0.2 nm),根据聚类的情况、结合能以及构象的合理性选取最佳构象进行分析并作为分子动力学模拟的初始构象,同时使用acpype程序[13]生成小分子配体的拓扑文件,用于构建分子动力学模拟中的复合物拓扑文件。
1.4 分子动力学模拟
本实验采用gromacs4.6.5软件,使用gromacs43 al力场[14],水分子采用SPCE模型[15],远程静电作用采用PME方法[16],系统的温度和压力通过V-rescale和Parrinel-Rahman实现控制[17]。将复合物置于一个立方体水盒子中,使用周期性边界条件以消除边界效应,加入离子中和电荷使整个体系处于中性环境,使用5000步最陡下降法进行能量最小化,然后进行400 ps的限制性动力学模拟,使得温度分别达到300 K,压力为1个大气压,温度和压力耦合系数为0.2 ps[18],待体系平衡后,进行50 ns的自由动力学模拟,模拟中采用的积分步长为2 ps,每2 ps保存一次轨迹结构用于后期数据的处理。通过分析复合物root mean squared deviation (rmsd)值,系统能量来判断复合物构象的稳定性。
1.4.1 底物和酶之间接触数和接触距离的计算
底物和葡聚糖酶之间的接触数和接触距离均采用GROMACS自带程序g_mindist进行计算,其中计算接触数时所用的截断距离为0.6 nm[19]。
1.4.2 复合物自由能计算
采用单轨迹方案的MM-PBSA方法计算葡聚糖酶与底物小分子的结合自由能,此方法是从复合物的MD模拟平衡的后5 ns轨迹中每隔一定的时间间隔取出体系结构,通过下式计算出平均结合自由能。
△Gbind=△Emm+△Gsol-T△S
其中△Gbind是结合自由能,△Emm、△Gsol和T△S分别是气相中的分子力学能、溶剂自由能和熵变对结合自由能的贡献。
△Emm=△Eele+△Evdw
式中△Eele和△Evdw分别表示气相中静电相互作用和范德华能。溶剂自由能也由如下的两部分组成:
△Gsol=△Gel+△Gnonel
式中△Gel和△Gnonel分别代表极性溶剂自由能和非极性溶剂自由能。前者的成分可以通过求解泊松—波尔兹曼方程获得[20],溶质和溶剂的介电常数分别设为1.0和80.0。后一项由下列的经验方程求解:△Gnonel=γSASA+β
式中△Gnonel代表非极性溶剂自由能,SASA代表溶剂可及接触面积,γ和β的值分别取为2.27 kJ/(mol·nm2)和3.85 kJ/mol。至于熵效应,可用简正模分析来计算,此项的计算非常耗时且对结果影响较小,因此一般情况下,可忽略熵变效应[21]。
1.4.3 氢键分析
本文中小分子配体和葡聚糖酶之间的氢键利用GROMACS自带程序g_hbond进行分析。当供体原子D和受体原子A之间的距离小于0.35 nm,且D-H-A之间的夹角大于120°时,即认为供体原子D和受体原子A之间形成氢键。
2 结果与分析
2.1 分子对接
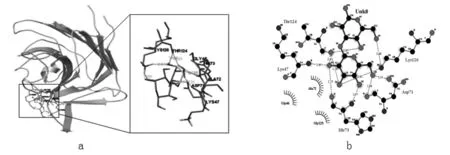
图1 复合物三维结构和平面图
通过利用分子对接方法,对二聚糖小分子配体与葡聚糖酶进行对接,选取自由能结合最小的构象,其试验结果如图1所示,图1a中,我们可以看出,Thr126、Thr124、Asp71、Ala72、His73、Gly46和Lys47残基参与二聚糖小分子的结合,并且其结合部位与Cheng等人研究的来自嗜热拟青霉的Ptlic16A晶体结合部位一致[5]。通过使用LigPlot软件[22],获得复合物结合部位平面图1b,从图中,我们可以看出,Ala72、Gly46和Gly125残基分别与小分子配体形成疏水相互作用,Thr124、Lys47、His73、Asp71和Lys126残基分别参与复合物氢键的形成。
2.2 分子动力学模拟
2.2.1 均方根偏差分析
从图2中,我们可以看出,葡聚糖酶和二聚糖分子结构从5 ns后趋于平衡,其中分别围绕着0.3 nm和0.15 nm值上下波动,在45~50 ns的时间内,二聚糖小分子结构的rmsd波动较小,并且系统总能量的平均波动量低于0.8%,因此可以认为系统在MD模拟后比较稳定,用此轨迹分析得出的结果是可靠的。
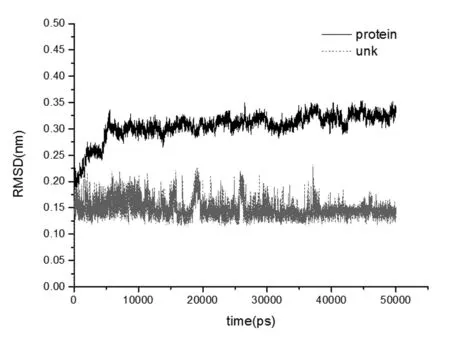
图2 酶和底物的RMSD值的变化
2.2.2 二聚糖和葡聚糖酶之间的接触数和分子间的距离
从图3A中我们可以看出,小分子配体与蛋白质之间的接触数在前10 ns内,随着时间的增加,其接触数目增多,在30~50 ns内,其值趋于平衡,总体波动范围为700~950,并且在794附件集中;图3B中为蛋白质与小分子之间的接触距离的频率分布,从图中我们可以看出,其值分布在0.1~0.3 nm之间,并且集中于0.22 nm,因此我们可以认为葡聚糖酶与二聚糖分子的结合较为紧密。

图3 A酶与底物的接触数,B酶与底物之间的距离
2.2.3 复合物结合自由能
通过上文对系统中RMSD的分析,我们选取了45~50 ns时间内的轨迹作为研究对象,每隔100 ps提取构象,共获得51个构象,通过利用MM-PBSA方法计算结合自由能,其结果如表1所示,从表1我们可以看出,小分子UNK与葡聚糖酶的结合自由能为-54.718 kJ/mol,这个结果表明小分子UNK与葡聚糖酶产生了比较强的结合能力。范德华作用能(△Evdw)为-88.473 kJ/mol,该结果表明范德华作用可以有效的促进小分子与蛋白质的结合。静电相互作用能(△Eele)和非极性溶剂自由能(△Gnonel)分别为-7.919 kJ/mol和-8.164 kJ/mol,这表明该作用为小分子的结合提供了较小的贡献。极性溶剂自由能(△Gel)为49.838 kJ/mol,这说明,该相互作用极大的衰减了静电相互作用,不利于小分子与蛋白质的结合。

表1 MM-PBSA计算所得的能量(kJ/mol)
2.2.4 活性位点氨基酸残基的贡献
为了进一步研究小分子UNK与葡聚糖酶分子间的相互作用模式,通过利用MM-PBSA程序计算每个氨基酸残基对结合小分子的贡献,试验结果如图4所示,Gly46、Leu63、Gln67、Tyr68、His73、Thr124、Gly125和Gln129对结合小分子的贡献较大,其中His73和Lys126残基可能作为亲核剂,为催化部位的活性位点,Gly46、Leu63、Gln67、Tyr68、Thr124、Gly125和Gln129可能与小分子结合识别有关。从表2 我们也可以看出,His73为碱性氨基酸,带有正电荷,与小分子之前形成的静电相互作用和范德华作用力较强,结合能为-4.4088 kJ/mol,Tyr68为疏水性氨基酸,极性溶剂自由能对静电作用影响较小,其对小分子的结合自由能为-4.6058 kJ/mol,其他氨基酸残基对小分子的结合自由能贡献均低于-1 kJ/mol。与上文对接试验结果较为一致。
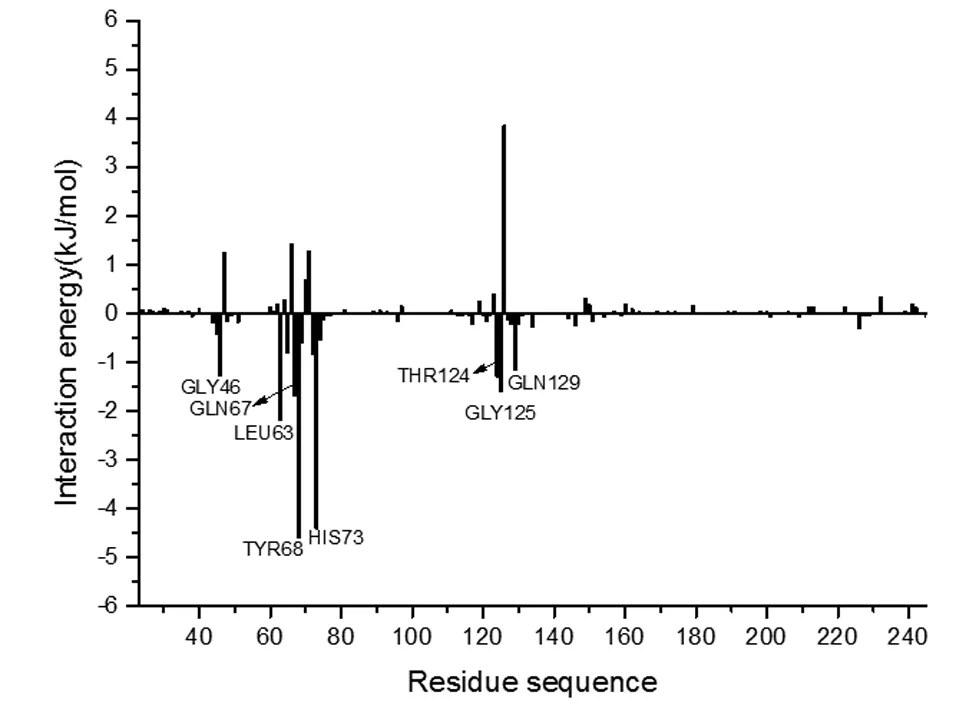
图4 底物与酶结合的相互作用谱

表2 能量分解的主要贡献残基各部分能量值
2.2.5 氢键分析
葡聚糖酶与小分子UNK之间形成的氢键数由图5所示,从图5可知,氢键数目随时间的变化而变化,在0~10 ns内,氢键数目呈递增的趋势,最大值为21;在20~40 ns时间段内,其氢键数目处于动态平衡,在3~15范围内波动;其整体的平均值为7。通过利用VMD软件分析,我们可以得出表3, 从表3我们可以看出His73和Thr124残基与小分子之间的氢键占有率均大于50%,这说明这些氨基酸残基与小分子之间形成的氢键较为稳定。

图5 底物与酶之间氢键数随时间的变化

表3 主要残基的氢键分析
3 结论
本文利用分子对接与动力学模拟的方法,研究葡聚糖酶结合底物的活性位点。通过分子对接技术,选取最佳的分子构象用于分子动力学模拟,用MM-PBSA方法计算了小分子UNK和葡聚糖酶之间的结合能,计算结果显示范德华相互作用、静电相互作用和非极性溶剂化自由能是形成复合物的主要驱动力,通过对各个氨基酸残基的贡献研究发现,Gly46、Leu63、Gln67、Tyr68、His73、Thr124、Gly125和Gln129是对小分子UNK的结合起关键作用的氨基酸残基。通过进一步研究小分子与葡聚糖酶之间的氢键数目及其对关键氨基酸的氢键分析,我们可以得出,小分子UNK与葡聚糖酶的结合较为紧密,同时也可以得出,His73和Thr124残基与小分子所形成的氢键较为稳定,对结合能贡献较大。
