基于风险管理的药品GMP 检查发起机制研究
2020-07-11曹嘉成张书卉焦灵利
曹嘉成,张书卉,焦灵利
江苏省食品药品监督管理局认证审评中心,南京210002
新修订的《中华人民共和国药品管理法》于2019 年12 月1 日起实施,规定取消对药品生产企业的《药品生产质量管理规范》(GMP)认证,改为对药品生产企业是否符合生产质量管理规范开展检查。《药品管理法》的这一变化,意味着GMP 检查由依企业申请变为监管部门依风险发起,如何科学地判断企业生产过程的风险,合理确定检查范围和检查频次,成为药品监管部门亟需思考解决的问题。药品生产监管风险较高,专业性较强,随着药品生产技术的日益精进,无菌保障要求不断提高,单抗、CAR-T 等新产品不断涌现,过去划片区的网格化监管,追求覆盖率的痕迹化监管已经不能满足新时代的监管形势,需要省级监管机构统筹谋划、统一尺度、加强风险研判,制定科学的GMP 检查发起机制,有针对性的开展检查,控制风险。
1 我国药品现行的GMP 检查发起机制
1.1 药品GMP 认证检查
《药品生产质量管理规范(2010 年修订)》于2011 年3 月1 日起正式开始施行,要求所有血液制品、疫苗、注射剂等无菌药品的生产应在2013 年12月31 日前达到GMP 要求,其他产品于2015 年之前全部达到GMP 要求。我国现行的GMP 按生产线进行认证,如一些企业有多条生产线,但对于企业本身而言,质量管理体系是一致的,同一剂型的不同车间设备及管理也几乎相近,这类企业往往都是大型企业,管理较完善,对于这种企业的反复认证是对行政资源的一种浪费。一些规模较小的企业,往往只有1~2 个主要品种,由于经济效益不高,这类企业往往人员素质相对较低,管理较薄弱,但这类企业只有到期换证才接受检查,中间空档期存在检查缺失的风险。
1.2 药品生产监督检查
在新版GMP 首轮基本完成后,监管部门意识到GMP 认证检查不能保持生产监管的高压态势,国家局和省局逐步加强了监督检查的力度,对部分企业组织飞行检查或跟踪检查,2016 年国家局对204家高风险企业进行了飞行检查[1],但相对于全国近6000家药品生产企业而言,只是冰山一角。各省级监管部门每年也会对辖区内的企业开展有针对性的检查,如江苏局依2019 年对57家重点企业进行了检查。
1.3 基于风险的检查发起机制
仅仅通过产品本身的风险开展检查,会造成一些综合性生产企业被多次、重复检查,对于合规情况较好的企业,频繁的检查不仅是对行政资源的浪费,也给企业造成了一定的负担。监管部门应根据企业的剂型风险,结合企业历史合规情况及近年来接受检查的情况,制定一套基于风险的检查发起机制,合理安排检查资源,对部分高风险的企业加大检查力度,同时应鼓励企业自律,对一直以来合规情况较好的企业,适当减少检查频次。
2 国外药品生产检查发起机制研究
2.1 美国药品生产检查发起机制
韩亮等[2]在《美国FDA 药品生产质量监管体系》中介绍,FDA 的药品生产检查主要分为3 种:一是新药和仿制药申请的批准前检查;二是常规GMP检查;三是有质量投诉或者突发事件等的有因检查。药品上市后的检查主要包括常规GMP 检查和有因检查,FDA 并没有明确规定现场检查的有效期,而会根据药品生产企业风险评估模型,确定检查发起的频次。FDA 建立的药品生产风险评估模型主要从产品本身、质量体系和设施设备情况、生产中操作不当三个方面分析风险要素[3],给其进行赋值,根据得分结果评价企业风险高低,对风险大的企业加强监督,而对风险小的企业则降低检查强度。
2.2 欧盟国家药品生产检查发起机制
欧盟的药品监管主要由European Medicines Agency(EMA)与各成员国自身的药品监管机构组成,EMA 主要负责药品的集中注册审批工作,各成员国的药品检查机构负责各自区域的GMP 检查工作[4]。在德国,食品、药品的日常监管基本都属于各地方政府的职权。各地对生产和销售企业的检查是不定期的,基本采取飞行检查的方式。检查的频率取决于两大因素:一是企业产品本身的风险程度,例如注射剂等无菌制剂就要比片剂等普通固体药品检查频率高;二是依据历次的合规情况,如一个企业前10 次检查均合规情况良好,以后对其检查的频率就会降低,反之就升高[5]。
2.3 澳大利亚药品生产检查发起机制
澳大利亚治疗产品管理局(Therapeutic Goods Administration,TGA),对澳大利亚药品和医疗器械的质量、安全、有效性和供应负责,联邦和州政府都设有药品监管机构,负责药品生产的日常检查。TGA制定检查频次主要依据企业产品风险和历年来合规检查情况,如无菌产品初始合规检查每24 个月一次,如果连续3 次合规情况良好,则36 个月检查一次[6]。
3 药品生产风险要素分析
基于国外风险评估的原则,综合考虑我国现阶段检查发起实际,药品生产风险评估模型主要分为三部分:一是企业自身风险;二是企业合规历史;三是接受检查频次。
3.1 企业自身风险
药品生产企业的风险主要来源于四个方面:一是企业经营的情况;二是企业生产品种的风险;三是企业人员素质;四是企业设施设备的情况。对上述风险涉及的具体风险指标进行分析,见表1。

表1 企业自身风险
3.2 企业合规历史 见表2。
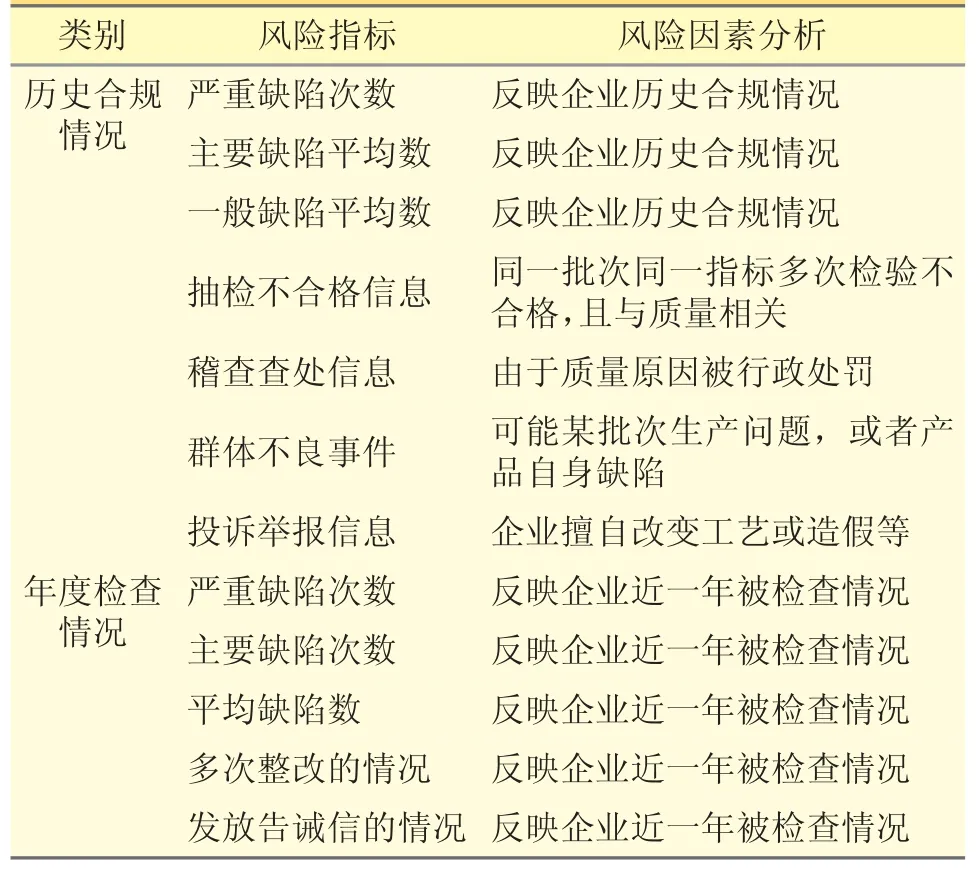
表2 企业合规历史
3.3 接受检查频次 见表3。
4 药品生产风险评估模型建立与试算
根据上述风险指标的分析,邀请业内资深检查员及企业质量管理人员对各风险指标进行分析,对风险指标的高低进行排序,根据排序情况对各指标进行合理赋值。赋值内容:企业自身风险;企业合规历史;接受检查频次及评分方法。
4.1 企业自身风险赋值 见表4。
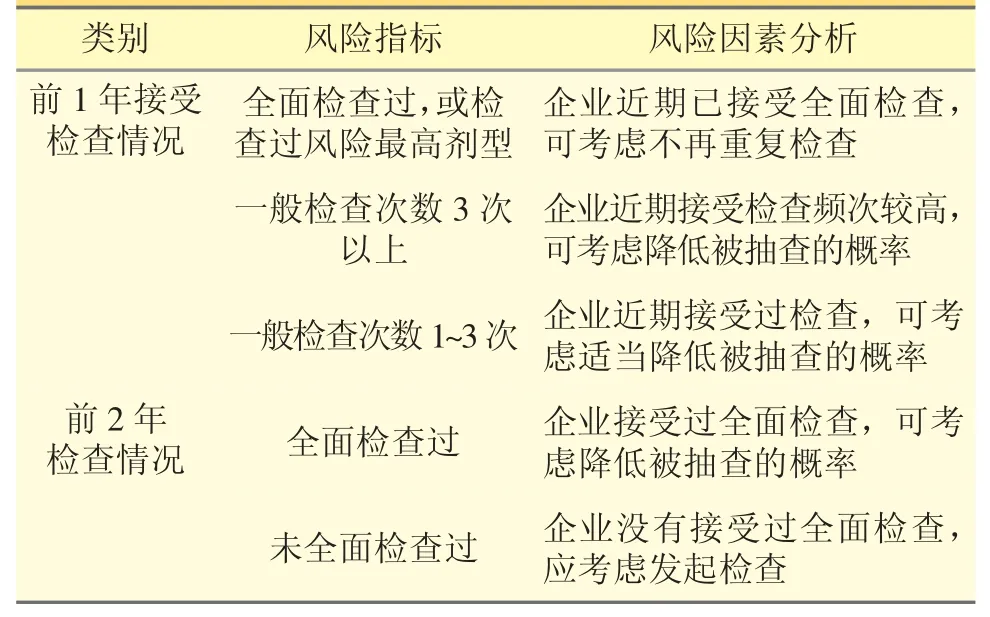
表3 接受检查频次
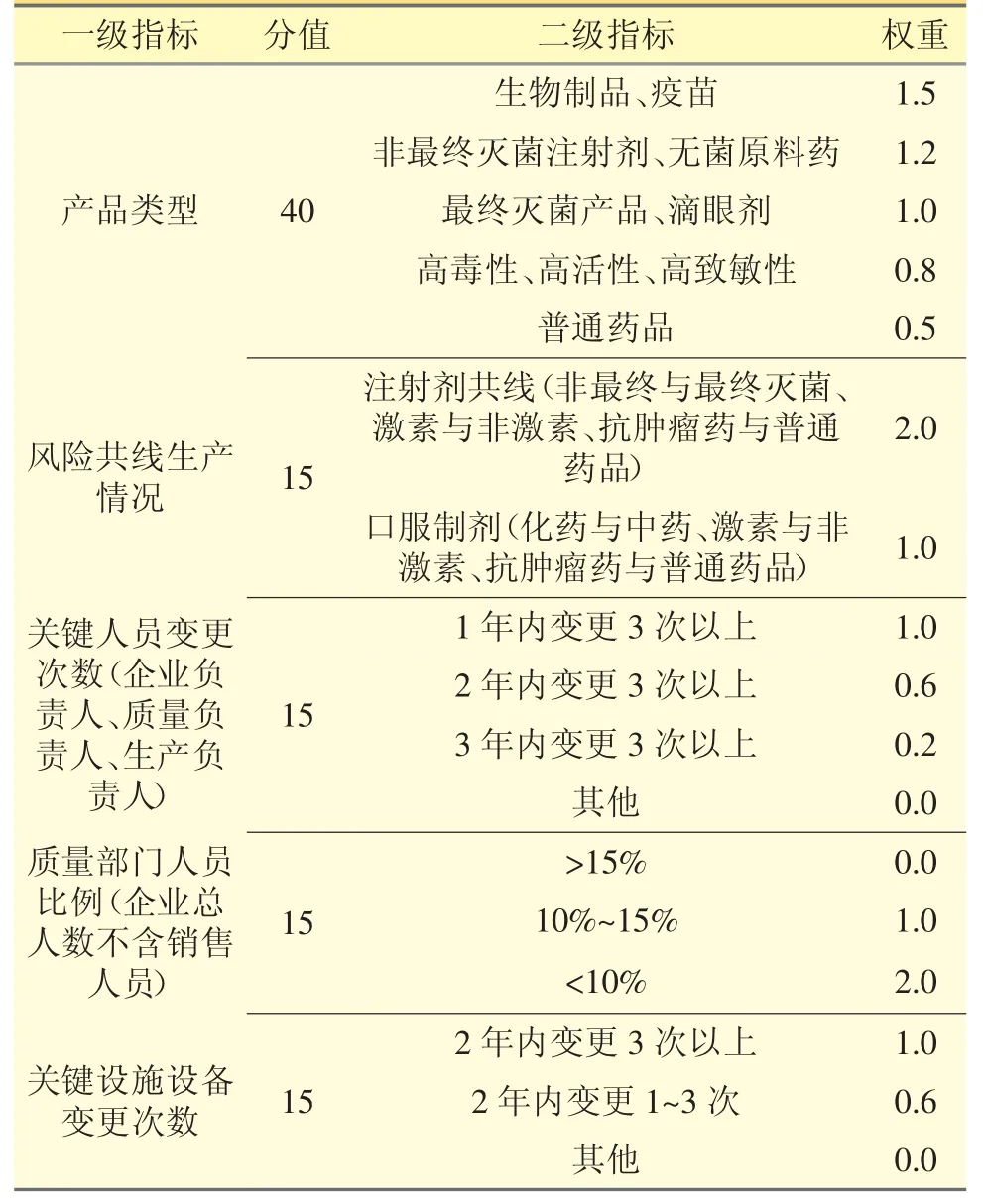
表4 企业自身风险赋值
4.2 企业合规历史赋值 见表5。
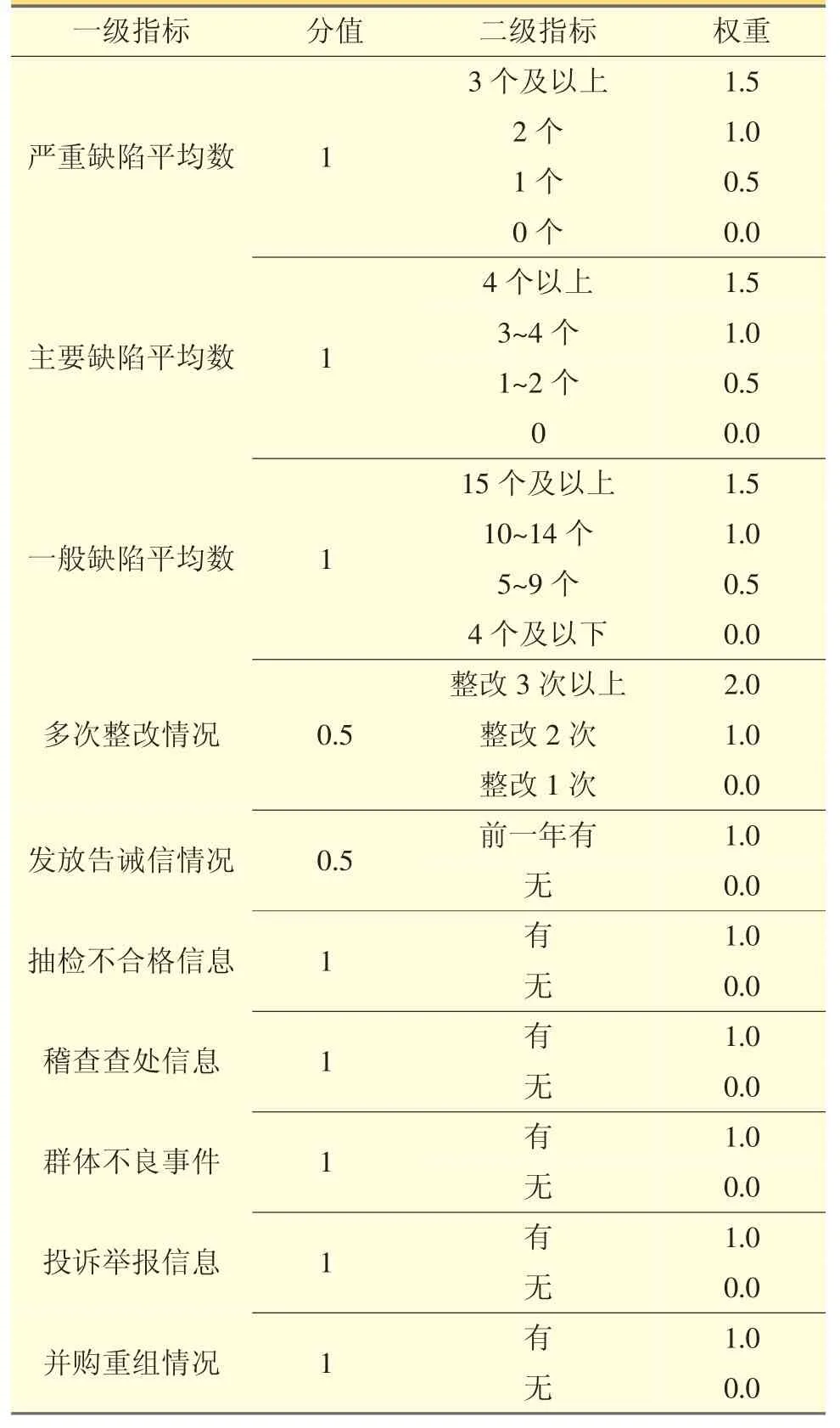
表5 企业合规历史赋值
4.3 接受检查频次赋值 见表6。

表6 接受检查频次赋值
4.4 评分方法
采用动态加权综合评价方法。将“企业自身风险”、“企业合规历史”、“接受检查频次”三个评价系统分别记为S1、S2、S3,每个系统的具体指标分为一级指标分值(X1、X2、…、Xm)和二级指标权重(P1、P2、…、Pm),打分=企业自身风险{∑(一级指标分值×二级指标权重)}×企业合规历史{∑(一级指标分值×二级指标权重)}×接受检查频次{∑(一级指标分值×二级指标权重)}。
4.5 风险评估模型试算
选取本省4家药品生产企业进行模型试算,见表7。
5 药品生产风险评估模型动态调整
药品生产企业的风险并不是固定不变的,企业的经营、品种、人员、设施设备等情况都在随时变化。风险指标的赋值主要根据监管需求和监管经验而定,赋值需要考虑多个因素:一是风险指标并不是孤立的,内在有着充分的联系,如企业经营情况不善,可能会导致人员流动性较大;二是风险具有两面性,如设施设备的更新可能会带来工艺改变或者验证不充分的风险,但是从长远考虑,是企业良性发展的必由之路;三是监管风险与企业风险的区分,如无菌制剂监管风险较高,但是如果企业无菌保障能力强,质量管理水平高,风险控制能力强,实际的监管风险并不高。生产风险指标的赋值不仅需要综合各方面因素,还要根据实际情况动态调整,既要根据监管经验,又要结合监管的新形势、新要求,同时还要考虑行业发展的动态。
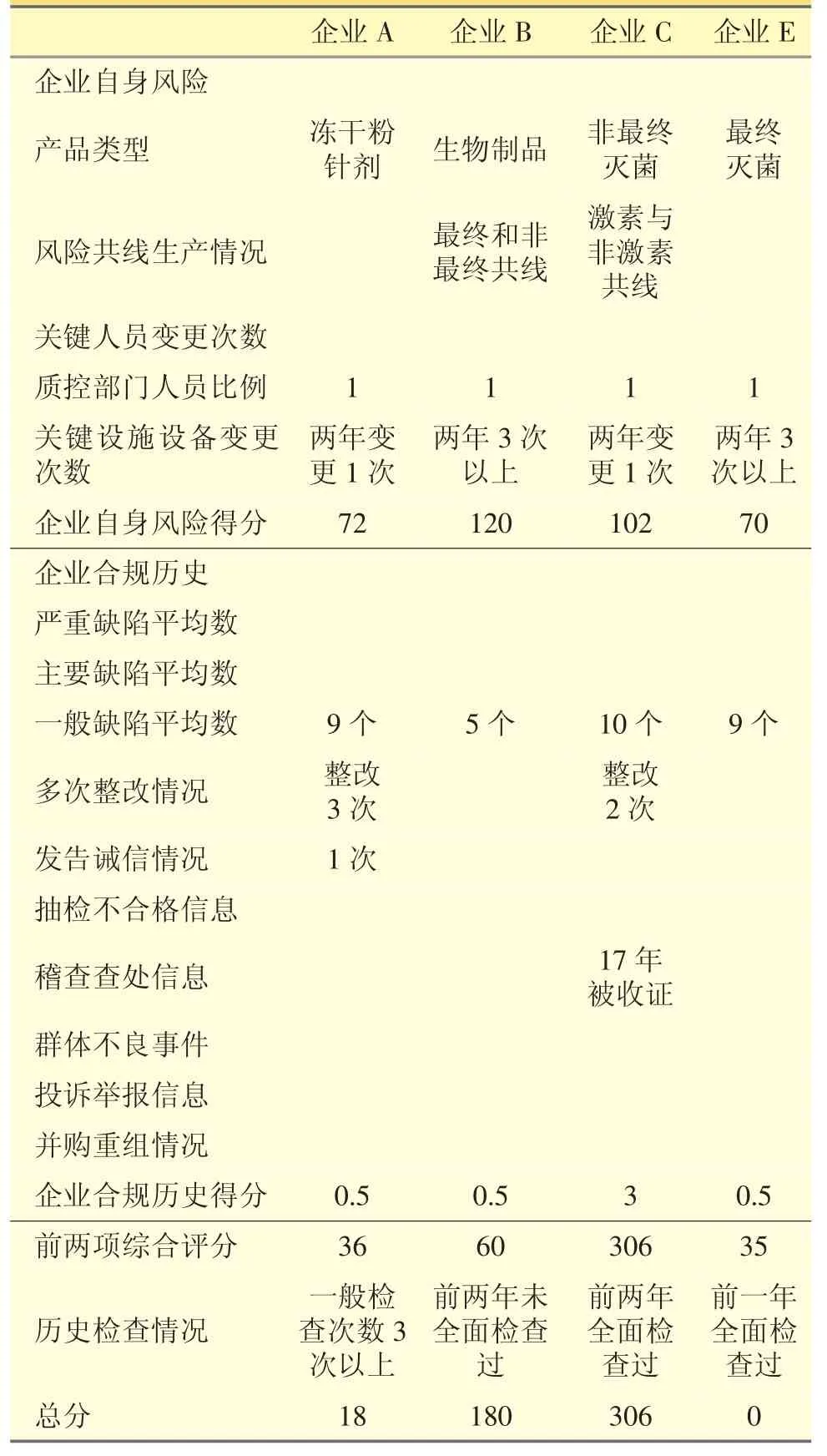
表7 江苏4家药品生产企业风险评估模型试算
监管风险指标赋值及权重应由资深检查员及监管人员根据检查经验和检查工作量拟定,通过检查的大数据反映的检查绩效来验证风险评估模型的合理性。同时,建立评分的动态调整机制,模型的评分根据检查绩效、监管需求、行业动态进行合理调整,保证风险评估模型的时效性、检查的指向性、监管的科学性。
6 结语
新修订的《药品管理法》取消了GMP 认证方式,由依照申请检查改为依照风险检查,本文利用风险评估工具及统计学手段,建立了药品生产风险评估模型,提高了GMP 检查的指向性,为GMP 检查新方式提供了科学依据,为合理利用行政资源提供有力抓手,为药品监管科学可持续发展、加快推动药品监管治理体系和治理能力现代化提供思路。
