RP-HPLC 法测定药用胶塞中16 种多环芳烃含量并作模拟提取研究
2020-07-11张晓芸刘露遥丁逸梅
张晓芸,刘露遥,丁逸梅
江苏省药物研究所有限公司,南京210009
多环芳烃(polycyclic aromatic hydro-carbons,PAHs)是一类由两个或两个以上苯环以稠环形式相连的有机化合物,主要包含萘、苊烯、芴、菲、蒽、荧蒽、芘、苯并蒽、艹屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚[1,2,3-cd]并芘、二苯并[a,h]蒽和苯并苝等。含有四到六个环的稠环多环芳烃具有致癌性,1976年国际癌症研究中心(IARC)公布了94 种动物致癌物质[1],其中多环芳烃化合物含有15 种,以苯并[α]芘为首个环境化学强致癌物质;多环芳烃具有很强的脂溶性,较难降解,并且容易在生物体内蓄积。到目前为止,欧盟2005/69/EC 指令(即76/769/EEC 之第27 次修订版)规定橡胶油中苯并[α]芘不得超过1mg·kg-1。2005 年9 月德国发布的《德国食品和商品法》中规定,PAHs 总量的最大允许限量是10 mg·kg-1,苯并[α]芘最大允许限量是1 mg·kg-1;美国和中国等各国均通过法律或法令对多环芳烃的含量有明确的限量要求[1,2]。橡胶中多环芳烃存在于胶塞生产原料橡胶油中,橡胶油作为一种软化剂可以使橡胶具有良好的弹性和韧性。注射剂等药物常见的包装材料为橡胶胶塞,如果胶塞中残留的PAHs 迁移至药液中会对患者造成危害;通常选择药用胶塞的材质分为:注射用冷冻干燥无菌粉末覆聚四氟乙烯/乙烯共聚物膜氯化丁基胶塞和注射用卤化丁基橡胶塞(氯化或者溴化)。目前,多环芳烃的检测技术主要有化学滴定法、高效液相色谱法、气相色谱-质谱联用法、气相色谱法、电化学法、分光光度法及热透镜光度法、拉曼光谱分析法等[3-13]。其中在环境保护领域较为常见的主要有高效液相色谱法、气相色谱-质谱联用法。本文采用RP-HPLC 法测定药用胶塞中16 种多环芳烃的含量,并作其迁移试验。
1 实验部分
1.1 仪器与材料
仪器:高效液相色谱仪LC-2010CHT(编号:C21255010791,日本岛津公司);电子分析天平(BSA224S,赛多利斯科学仪器(北京)有限公司);手提式压力蒸汽灭菌器(上海划线医用核子仪器有限公司,编号:428)。
16 种多环芳烃混合对照品(PAHs-Solution16-22),来源:O2Si,批号:320418,质量浓度(萘1002 mg·L-1,苊2002 mg·L-1,茐200.1 mg·L-1,二氢苊999.9 mg·L-1,菲100.2 mg·L-1,蒽100.2 mg·L-1,荧蒽200.1 mg·L-1,芘99.84 mg·L-1,艹屈100.6 mg·L-1,苯并[a]蒽100.4 mg·L-1,苯并[b]荧蒽200.1 mg·L-1,苯并[k]荧蒽100.4 mg·L-1,二苯并[a]芘100.7 mg·L-1,二苯并[ah]蒽199.9 mg·L-1,苯并[ghi]苝200 mg·L-1,茚并[1,2,3-cd]芘99.96 mg·L-1)。16 种多环芳烃混合对照品(PAHs-Mix-EPA610),来源:O2Si 标准品公司,批号:300821,质量浓度均为200 μg·mL-1;萘对照品,来源:阿拉丁试剂公司,批号:C1819049,纯度98%。
药用胶塞:①注射用冷冻干燥无菌粉末用覆聚四氟乙烯/乙烯共聚物膜氯化丁基胶塞,来源:江苏华兰药用新材料股份有限公司,批号:M170406122-78;②药用镀聚四氟乙烯膜氯化丁基橡胶/聚异戊二烯橡胶塞,来源:新加坡西式西药服务公司,批号:3138 010040;③注射用卤化丁基橡胶塞(溴化),来源:江苏博生医用新材料股份有限公司,批号:16 102362;④注射用冷冻干燥无菌粉末氯化丁基橡胶塞,来源:江苏华兰药用新材料股份有限公司,批号:170508159-60。
药品与试剂:注射用兰索拉唑,来源:江苏某药业股份有限公司,批号:180301,180302,180303;注射用帕瑞希布钠,来源:杭州某药业有限公司,批号:19010814,19010914,19 011014;注射用万古霉素,来源:浙江某药业股份有限公司,批号:20170901,20170902,20 170903;乙腈,来源:Honeywell,批号:S3ZA1H);乙醇、正己烷为分析纯;实验用水为重蒸水。
1.2 方法
1.2.1 色谱条件 色谱柱:Kromasil 100-5-C18(4.6 mm×250 mm,5 μm);流速:1.2 mL·min-1;柱温:35 ℃;检测波长:220(萘、苊、二氢苊)、254(茐、菲、蒽、荧蒽、芘、艹屈、苯并[a]蒽、苯并[b]荧蒽、苯并[k]荧蒽、二苯并[a]芘、苯并[ghi]苝)、295(二苯并[ah]蒽、茚并[1,2,3-cd]芘)nm;进样量:10 μL。流动相:色谱纯乙腈、屈臣氏蒸馏水(采用梯度洗脱:0~27 min,乙腈65%;27~41 min,乙腈65%~100%;41~43 min,乙腈100%~65%;43~55 min,乙腈65%)。16 种PAHs 对照品的典型图谱见图1。
1.2.2 气相色谱条件 Thermo Scientific Trace GC Ultra-ISQ 气质联用仪,软件:Xcalibur;离子源:EI;色谱柱:Agilent DB-5 MS UI(30m×250μm×0.25μm);进样量:1.5 μL;不分流;恒定流量:1.2 mL·min-1;进样口温度:280 ℃;柱温:60 ℃,保持1 min,以10 ℃·min-1升至110 ℃,保持2 min,以5 ℃·min-1升至300 ℃,保持10 min;传输线温度:250 ℃;离子源温度:250 ℃;溶剂延迟:6.0 min;定量离子搜索模式(SIM)。定性与定量离子选择见表1。
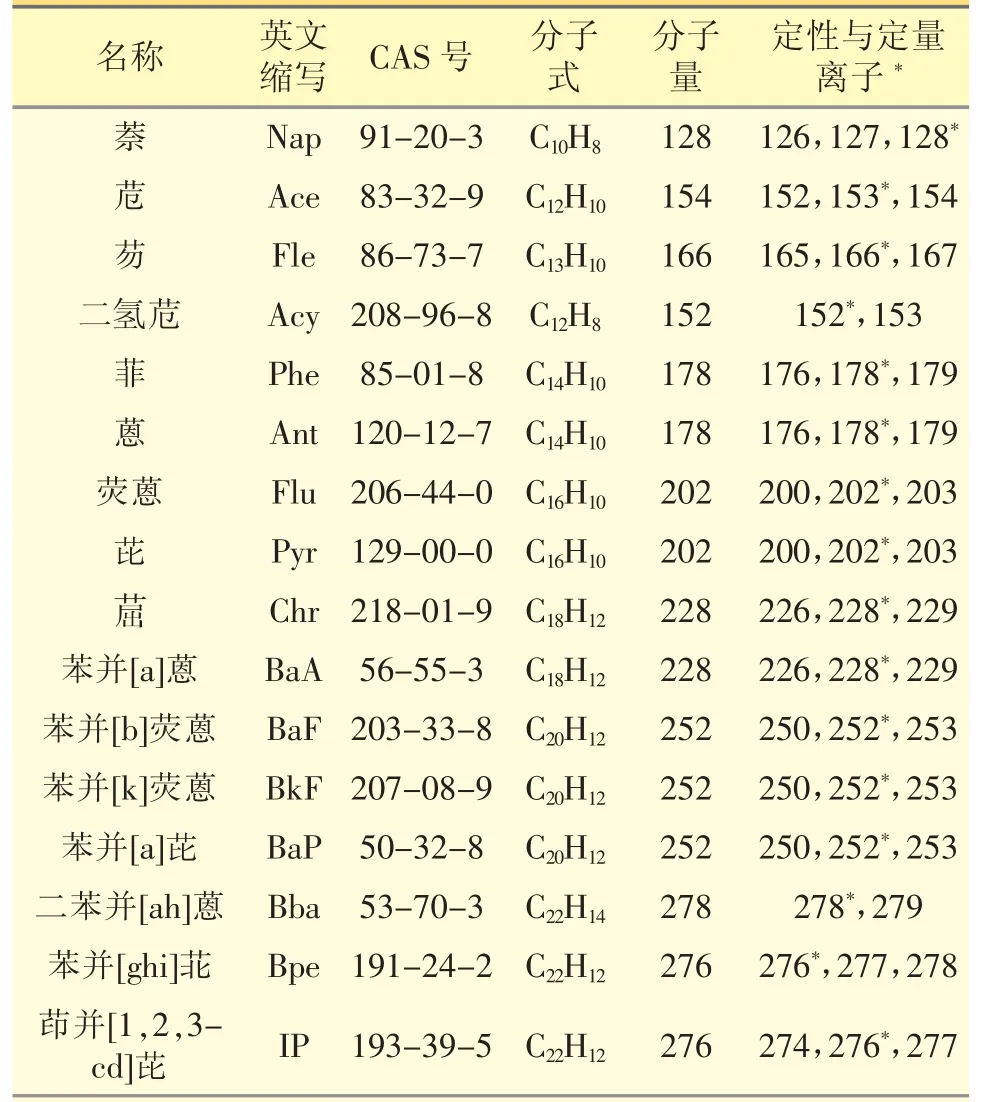
表1 定性与定量离子选择
1.2.3 提取试验 胶塞经121 ℃高压灭菌15 min后,取出剪碎,称取约10 g,精密称定,置100 mL 锥形瓶中,加20 mL 无水乙醇,50 ℃超声4 h;取出冷却,取上清液即为提取液。
1.2.4 迁移试验
1.2.4.1 对照品溶液制备 精密量取PAHs 混合对照品适量,至100 mL 量瓶中,用甲醇稀释成PAHs含量为0.1 μg·mL-1的混合对照品溶液,待用。
1.2.4.2 样品溶液制备 ①取注射用兰索拉唑(批号:180301,180302,180303)冻干粉末3 瓶,模拟临床使用,每瓶加入5 mL 0.9% NaCl,混合均匀;精密量取10 mL 该样品溶液,精密加入5 mL 正己烷,涡旋5 min,静置分层,精密量取有机层1.0 mL,用高纯氮气吹干后,加入1.0 mL 无水乙醇复溶。②取注射用帕瑞希布钠(批号:19010814,19010914,19011014)冻干粉末,模拟临床使用每瓶加入5mL 0.9% NaCl,取2 瓶混合均匀后备用;精密量取5.0mL 该样品溶液,加入5 mL 正己烷,涡旋5 min,静置分层,精密量取有机层1.0mL,用高纯氮气吹干后,精密加入1.0mL无水乙醇复溶。③取注射用万古霉素(批号:20170901,20170902,20170903)冻干粉末,模拟临床使用每瓶加入5 mL 0.9% NaCl,取2 瓶混合均匀后备用;精密量取5.0 mL 该样品溶液,加入5.0 mL 正己烷,涡旋5 min,静置分层,精密量取有机层1.0 mL,用高纯氮气吹干后,精密加入1.0 mL 无水乙醇复溶。
1.3 结果
1.3.1 提取实验
1.3.1.1 线性范围 精密量取PAHs 混合对照品溶液适量,用乙腈逐级稀释,制备线性溶液,浓度分别为0.1000、0.500、1.000、5.000、10.00 μg·mL-1;精密量取上述对照品溶液10 μL,注入液相色谱仪测定,以对照品浓度为横坐标,峰面积为纵坐标,绘制标准曲线。结果表明,各PAHs 在0.1000~10.00 μg·mL-1范围内线性关系良好,相关系数(r)≥0.999。以信噪比S/N=3 为最低检出限,检出限0.751 5 ng·mL-1~24.96 ng·mL-1;以信噪比S/N=10 为最低定量限2.505 ng·mL-1~83.20 ng·mL-1。见表2。
1.3.1.2 进样精密度试验 精密量取对照品溶液10 μL,注入液相色谱仪测定,连续进样5 次,测得峰面积的RSD 均≤2.0%,表明进样精密度良好。
1.3.1.3 回收率实验 取“1.2.3”项的胶塞10 g,共9份,使用含有浓度为0.1000、0.5000、1.000μg·mL-1的PAHs 对照品溶液20mL,按照“1.2.3”项下方法制备胶塞样品溶液,精密量取该溶液10μL,注入液相色谱仪测定,以外标法计算得到回收率为88.74%~99.27%。
1.3.1.4 重复性试验 取“1.2.3”项的胶塞10 g,共6 份,精密加入浓度为0.5000 μg·mL-1LPAHs 混合对照品溶液20 mL,按照“1.2.3”项下方法制备胶塞样品溶液,精密量取该溶液10.0 μL,注入液相色谱仪测定,测得的RSD≤3.0%。
1.3.1.5 胶塞提取 对市售的药用胶塞进行PAHs的检测,每种胶塞按照“1.2.3”项下方法制备胶塞样品溶液。萘、苊和二氢苊的检测波长为220 nm,依法测定,由图2A 可知,胶塞中可能存在萘,提取液图谱中苊的保留时间与对照品图谱中的苊的保留时间不一致,待GCMS 进行分子量鉴定;茐、菲、蒽、荧蒽、芘、艹屈、苯并[a]蒽、苯并[b]荧蒽、苯并[k]荧蒽、二苯并[a]芘和苯并[ghi]苝的检测波长为254 nm,由图2B 可知,该类化合物均未检出;二苯并[ah]蒽和茚并[1,2,3-cd]芘的检测波长为295 nm,由图2C 可知,这两种化合物均未检出。其提取液HPLC 图谱见表3。
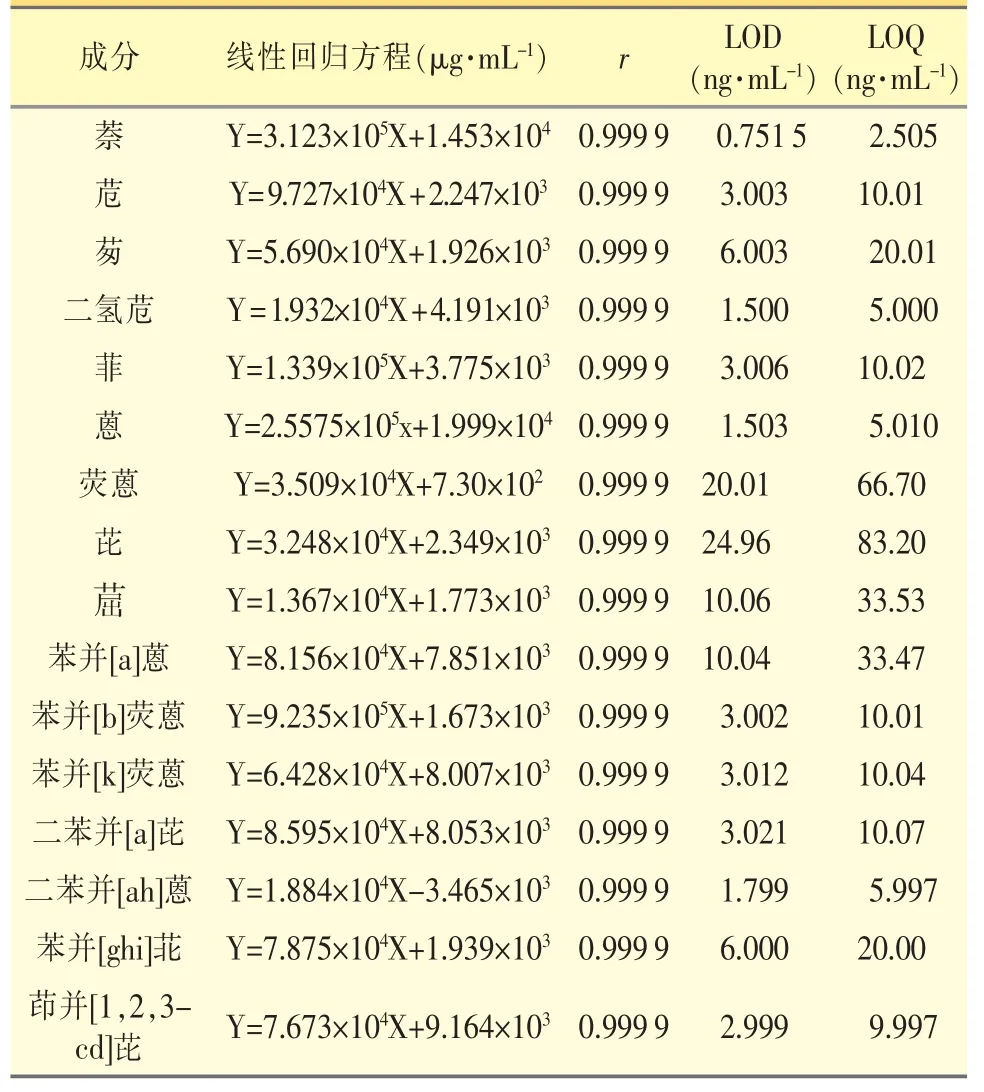
表2 16 种PAHs 的回归方程
1.3.2 迁移实验
1.3.2.1 萃取溶剂筛选 本实验筛选了溶剂:乙醚、乙酸乙酯、二氯甲烷、三氯甲烷、正己烷,乙醚沸点太低,难以精密量取;乙酸乙酯、二氯甲烷、三氯甲烷对主药部分溶解,干扰多环芳烃的检测;多环芳烃在正己烷中溶解性较好,主药及杂质在正己烷中溶解性较差,使用正己烷提取干扰较少,回收率高。
1.3.2.2 回收率试验 分别取注射用兰索拉唑样品(批号180301)、注射用帕瑞昔布钠(批号19010814)和注射用万古霉素样品(批号20170901),精密加入含PAHs 浓度为0.1000、0.5000、1.000 μg·mL-1的乙醇溶液,按照“1.2.4.2”方法制备样品溶液;精密量取该溶液10 μL,注入液相色谱仪,记录各色谱峰峰面积,采用外标法计算回收率及RSD 值,注射用兰索拉唑平均加样回收率为94.28%~98.81%,RSD≤5.0%;注射用帕瑞昔布钠平均加样回收率为93.61%~99.43%,RSD≤5.0%;注射用万古霉素平均加样回收率为93.34%~98.89%,RSD≤5.0%。见表4。

表3 胶塞多环芳烃提取结果(ng·g-1)
1.3.2.3 注射液中PAHs 的测定 ①选取3 批注射用兰索拉唑样品(批号:180301,180302,180303),进行PHAs 迁移试验,按照“1.2.4.2”项下方法制备样品溶液;精密量取该溶液10.0 μL,注入液相色谱仪测定,3 批样品中均未检出PHAs。②取3 批注射用帕瑞希布钠(批号:19010814,19010914,19011014),进行PHAs 迁移试验,按照“1.2.4.2”项下方法制备样品溶液;精密量取该溶液10.0 μL,注入液相色谱仪测定,3 批样品中均未检出PHAs。③取3 批注射用万古霉素(批号:20170901,20170902,20170903)进行PHAs 迁移试验,按照“1.2.4.2”项下方法制备样品溶液;精密量取该溶液10.0 μL,注入液相色谱仪测定,3 批样品中均未检出PHAs。
2 结论
本实验使用高效液相色谱法测定药用胶塞中16 种PAHs,各PAH 范围在0.100 0~10.00 μg·mL-1浓度之间内,其峰面积与浓度线性关系良好,相关系数r≥0.999;检测限0.751 5~24.96 ng·mL-1;定量限2.505~83.20 ng·mL-1;精密度的RSD≤2.0%;重复性的RSD≤3.0%;平均加样回收率为88.74%~99.27%。
经GCMS 定性分析,表明胶塞中提取物为萘,目前GCMS 也能够将PAHs 良好分离,且灵敏度良好;但考虑GCMS 自身的缺陷性,适用于定性和半定量分析,且GCMS 相对成本较高。本文仍采用液-液萃取的方式,以HPLC-UV 法考察PAHs 的迁移实验。
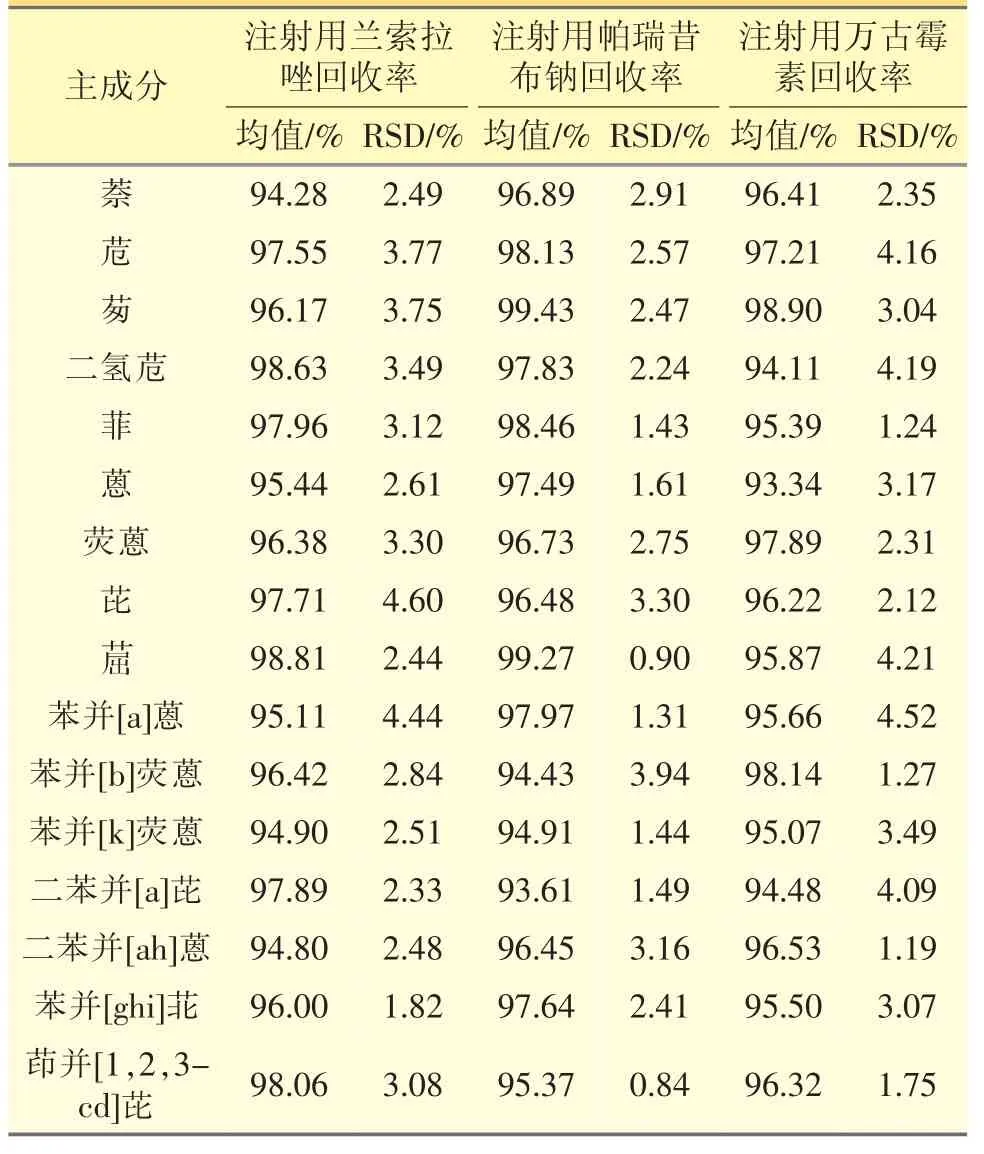
表4 注射用兰索拉唑、帕瑞昔布钠和万古霉素中PAHs 的迁移平均回收率及RSD 值
本试验选用注射用兰索拉唑等3 个样品为模型注射剂,建立了16 种PAHs 的迁移实验,通过加标方式考察该方法的专属性,注射用兰索拉唑平均加样回收率为94.28%~98.81%,RSD≤5.0%;注射用帕瑞昔布钠平均加样回收率为93.61%~99.43%,RSD≤5.0%;注射用万古霉素平均加样回收率为93.34%~98.90%,RSD≤5.0%。本试验采用的HPLC法测定药用胶塞中的PAHs,方法良好,能够快捷、简便、准确地检测出橡胶胶塞中16 种PAHs 的含量,适用于橡胶胶塞中PAHs 的测定。通过溶剂筛选得到正己烷为注射剂药液的合适的萃取溶剂,能够准确考察注射液中16 种PAHs 的迁移。
