TAK1/NF-κB在小鼠心肌肥厚中的变化及意义
2020-07-03唐其柱沈涤非卞洲艳
周 恒 唐其柱 沈涤非 严 玲 卞洲艳 袁 园
心肌肥厚是心脏负荷增加或遭受损伤时的一种代偿性反应,虽然在早期可以减轻心室壁压力和维持心排出量,但长期持续的心肌肥厚会发展为失代偿,导致心室功能障碍,最终发生心力衰竭[1]。压力负荷是心肌肥厚最常见的原因之一,高血压、主动脉瓣狭窄等疾病均会导致左心室压力负荷的升高,促进心肌肥厚发生。心肌肥厚涉及心肌细胞增大和功能障碍、间质纤维化等多种病理生理反应,参与心肌肥厚的分子机制也非常复杂,目前尚未完全阐明。虽然针对神经体液机制的治疗能够在一定程度上改善心肌肥厚,但是仍有相当数量的心肌肥厚与心力衰竭患者疗效不佳[2]。探索心肌肥厚的发生、发展机制,寻找潜在的防治靶点,对于减少高血压等心血管疾病的并发症、遏制心力衰竭的发展具有重要意义。
近年来研究显示,炎性反应与心肌肥厚及心力衰竭关系密切,可能成为调节心肌肥厚进程的新靶点[3]。转化生长因子活化激酶1(transforming growth factor-β activated kinase 1, TAK1)是促炎细胞信号转导通路中的关键分子,能够激活下游的核因子(nuclear factor, NF)-κB,促进炎性细胞因子释放,介导炎性反应发生[4]。然而,TAK1在心肌肥厚中的作用尚不明确。本研究拟通过胸主动脉缩窄术(aortic banding, AB)建立压力负荷诱导的心肌肥厚模型,检测心肌组织中TAK1/NF-κB与炎性反应水平,以探索TAK1/NF-κB介导的炎性反应在心肌肥厚中的作用,为防治心肌肥厚与心力衰竭提供新的潜在分子靶点。
材料与方法
1.实验动物:以8~10周龄、体质量23.5~27.5g的雄性野生型C57BL/6J小鼠为研究对象。小鼠饲养于SPF级动物中心的独立送回风净化笼具(IVC)中,饮用水经高压处理,饲料经60Co照射灭菌。系统温度维持于22~24℃,湿度为50%~60%, 12h明暗交替。小鼠随机分为两组:假手术对照(Sham)组与心肌肥厚模型(AB)组。
2.心肌肥厚模型的建立:采用胸主动脉缩窄术建立小鼠心肌肥厚模型。腹腔注射3%戊巴比妥钠麻醉小鼠,沿第2~3肋间水平切开皮肤,分离肌肉及软组织,游离胸主动脉,以7-0手术缝线横穿主动脉下方,将去尖的26G(小鼠体质量25.0~27.5g)或27G(小鼠体质量23.5~25.0g)注射器针头平行放置于血管上方,针头连同主动脉一起结扎,随后立即抽出针头,使用超声多普勒检测评估主动脉狭窄程度,以确定手术造成了主动脉约70%狭窄[5]。对照组在分离出主动脉后,只挂线不结扎,其余手术步骤与AB组相同。术后8周进行各项指标检测。
3.超声心动图:异氟烷(1.5%~2.0%)持续吸入麻醉,麻醉状态稳定后,小鼠左侧卧位,采用高频超声诊断仪,频率为15MHz。取胸骨旁左心室乳头肌水平短轴切面,测量舒张期室间隔厚度(IVSD)、舒张期左心室后壁厚度(LVPWD)、左心室收缩末期内径(LVESD)、左心室舒张末期内径(LVEDD)以及短轴缩短率(FS)等指标。
4.心脏取材:称量小鼠体质量,采用颈椎脱臼法处死小鼠,立即取出心脏,称量心脏质量。取材完成后半数心脏放至-80℃冰箱保存,用于Western blot法与RT-PCR法检测。其余心脏放入4%甲醛溶液中固定,用于病理学与免疫荧光检测。
5.病理学检测:将固定后的小鼠心脏横切,进行脱水、透明、浸蜡,包埋入石蜡块。制备出4~5μm厚的组织切片,进行常规HE与天狼猩红染色,显微镜下观察、拍照。使用图像分析软件(Image Pro-Plus 6.0)计算心肌细胞横截面积与心肌组织胶原容积分数。
6.免疫荧光:组织切片采用柠檬酸盐高压修复法进行抗原修复,10%羊血清37℃孵育60min进行封闭。CD68一抗4℃孵育过夜,绿色荧光标记的二抗湿盒中37℃孵育60min。滴加含有DAPI的封片剂封片,放置5min左右后,在荧光显微镜下观察、拍照。拍照时在同一视野下分别拍摄绿色激发荧光与蓝色激发荧光两张图片,并使用Image-pro plus 6.0软件合成,计算各组CD68阳性细胞数。
7.实时定量RT-PCR法:TRIzol提取心肌组织总RNA,分光光度法检测RNA纯度及浓度。每个样本取2μg总RNA,使用反转录试剂盒(20μl反应体系)进行反转录。得到的cDNA以LightCycler 480 SYBR Green 1 Master Mix反应体系进行PCR扩增:95℃变性10min,随后95℃ 15s进行40个循环,GAPDH作为内参。
8.Western blot法:使用RIPA裂解液研磨心肌组织,BCA蛋白定量试剂盒进行蛋白定量。蛋白样品经SDS-PAGE进行分离,然后转移至PVDF膜,于封闭液中封闭2h。继以待测蛋白的一抗4℃孵育过夜,荧光标记的二抗室温孵育1h,置于双通道荧光扫描仪中扫膜,计算机分析结果。

结 果
1.超声心动图与心脏质量:AB术后8周的模型组小鼠左心室室壁厚度与室腔大小明显增加,而短轴缩短率明显下降,出现了明显的左心室结构异常与心功能障碍(表1)。心肌肥厚模型组小鼠心脏质量/体质量(HW/BW, mg/g)较对照组明显增加(4.07±0.12mg/g vs 8.07±0.45mg/g,P<0.05)。
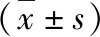
表1 两组小鼠超声心动图检测结果
与Sham组比较,*P<0.05
2.病理学检测结果:HE染色显示,模型组小鼠心肌细胞排列紊乱,横截面积较对照组明显增大,出现了明显的心肌肥厚(图1)。PSR染色显示,模型组小鼠组织间隙染为红色的胶原成分明显增多,心肌组织胶原沉积、纤维化(图2)。

图1 心肌组织HE染色结果(×400)A.Sham组;B.AB组

图2 心肌组织PSR染色结果(×400)A.Sham组;B.AB组
3.肥厚心肌组织中炎性反应增加:通过CD68免疫荧光标记单核-吞噬细胞,检测心肌组织炎性浸润情况。心肌肥厚模型组小鼠心肌组织中CD68阳性细胞数明显多于对照组(图3)。此外,RT-PCR检测发现,心肌组织肿瘤坏死因子-α(TNF-α)与白介素-1β(IL-1β)等炎性反应标志物的mRNA表达水平在模型组中明显升高(图4)。

图3 心肌组织CD68免疫荧光检测(×400)
4.肥厚心肌组织中TAK1/NF-κB通路激活:Western blot法检测结果显示,心肌肥厚模型组小鼠心肌组织中TAK1、κB抑制蛋白激酶β(IKKβ)与κB抑制蛋白α(IκBα)的磷酸化水平较对照组明显上调,IκBα总蛋白水平降低,NF-κB p65磷酸化水平增加,TAK1/NF-κB信号通路的激活(图5)。
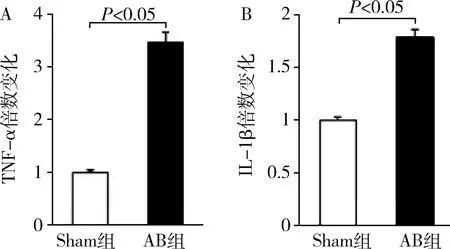
图4 心肌组织炎性反应标志物表达结果A.TNF-α;B.IL-1β

图5 肥厚心肌组织中TAK1/NF-κB信号通路的激活
讨 论
心肌肥厚是心力衰竭的关键性临床阶段,是多种心血管疾病向心力衰竭发展的共有病理生理过程,涉及心肌细胞增大、排列紊乱和功能障碍、间质纤维化等多种病理改变。本研究通过主动脉缩窄术成功建立了压力负荷诱导的心肌肥厚模型,并使用超声心动图、HE与PSR染色进行评估,在术后8周观察了模型组小鼠心肌肥厚、室腔扩大、心功能不全以及各种病理学表现。
然而,参与心肌肥厚的分子机制非常复杂,迄今尚未完全阐明。近年来,炎性反应及其相关信号通路在心肌肥厚中的作用逐渐受到重视。临床研究显示,左心室肥厚患者体内存在系统性炎性反应,其血清中的炎性反应标志物 CRP 以及促炎性细胞因子TNF-α、IL-1β与IL-6的表达均明显升高[6]。另外,心肌组织中包括心肌细胞在内的多种细胞能够分泌炎性细胞因子,并表达炎性细胞因子受体;心脏既是炎性细胞因子产生的场所,也是其作用的靶器官[7]。因此,炎性反应不仅仅是心肌肥厚与心力衰竭的标志物与预后指标,同时参与了心肌肥厚的进程,与心肌细胞肥大与心肌间质纤维化等病理过程密切相关[8]。本研究显示,心肌肥厚模型组小鼠心肌组织中CD68阳性细胞数明显增多,TNF-α与IL-1β等炎性反应标志物的mRNA表达水平也明显升高,提示心肌出现炎性细胞浸润与促炎性细胞因子的产生。
NF-κB是调节炎性反应的关键转录因子,在细胞生长、增殖、凋亡和细胞外基质重塑等多种病理生理过程中也发挥重要作用[9]。NF-κB二聚体(经典组合为p65和p50)通过与NF-κB抑制剂(IκBs)结合而失活,IκBs在静息状态的细胞胞质维持NF-κB的稳定[10]。而在应激情况下,IκBs可被IκB激酶(IKKs,如IKKβ)磷酸化,导致IκBs的泛素化并降解,解除IκBs对NF-κB的抑制,进而引起NF-κB的释放和活化[11]。已有研究发现,在体外培养的大鼠心肌细胞中,NF-κB可被多种肥厚刺激物激活,包括血管紧张素Ⅱ、苯肾上腺素和内皮素-1[12]。本研究结果显示,AB组IKKβ与IκBα的磷酸化水平较对照组明显上调,IκBα总蛋白水平降低,NF-κB p65磷酸化水平增加,进一步证实NF-κB信号通路可在压力负荷诱导的心肌肥厚中激活。
但是,NF-κB在心肌肥厚中的作用尚存争议。过表达NF-κB会促进肥厚刺激引起的心房利钠肽的表达和心肌细胞的增大,而超抑制型IκBα突变体或显性负性IKKβ突变体的表达会抑制NF-κB并减轻这些肥厚[12]。小鼠心脏特异性p65缺失可缓解压力负荷诱导的心肌肥厚,改善病理性心肌重构,增强心脏收缩功能[13]。以sh-p65 RNA转染心脏,抑制NF-κB,可减轻Myo转基因小鼠的自发性心肌肥厚[14]。这些发现提示NF-κB具有促进心肌肥厚的作用,然而,也有一些研究存在相反的结果。Hikoso等[15]报道,心脏特异性敲除IKKβ会降低NF-κB活性,但却加重了压力负荷诱导的心肌肥厚、心室扩张以及心功能障碍。NF-κB必须调节蛋白(NF-κB-essential modulator, NEMO)是IKK复合物的调节亚基,通过其心脏特异性缺失可抑制NF-κB通路,并促进压力负荷诱导的心肌肥厚与心力衰竭[16]。这些结果则提示NF-κB通路对心肌肥厚也具有保护作用。NF-κB在心肌肥厚中作用不一致的原因可能是由于模型构建的不同、实验环境的不同、NF-κB作用靶点的多样性以及NF-κB上游调控分子的多样性所导致的。
TAK1是一种丝氨酸/苏氨酸激酶,能够被转化生长因子-β、Toll样受体、NOD样受体等多种因素激活,是NF-κB信号通路关键的上游调节因子[17]。TAK1可以使IKK磷酸化,激活IKK复合物,促进IκB的磷酸化与降解,致使NF-κB的游离及活化入核,诱导TNF-α等促炎性细胞因子表达[18,19]。但是,心肌肥厚过程中TAK1的作用及其与NF-κB和炎性反应的关系,尚不明确。本研究在心肌肥厚小鼠模型中发现,TAK1的磷酸化水平较对照组明显增加,与NF-κB p65的变化趋势一致,同时伴有心肌组织炎性浸润,提示TAK1可能作为上游分子激活NF-κB以及炎性反应,并参与心肌肥厚的发生、发展。
综上所述,本研究在压力负荷诱导的小鼠心肌肥厚模型中,发现了TAK1/NF-κB通路的激活与心肌组织炎性浸润,提示TAK1/NF-κB及其介导的炎性反应参与了心肌肥厚的发生、发展,为探索心肌肥厚的分子机制与寻找潜在的防治靶点提供了一定的理论基础。
