靶向宿主的抗流感病毒药物研究进展
2020-06-19王萌颜海燕李玉环于莲
王萌,颜海燕,李玉环,于莲
·综述·
靶向宿主的抗流感病毒药物研究进展
王萌,颜海燕,李玉环,于莲
154007 黑龙江,佳木斯大学药学院药剂室(王萌、于莲);100050 北京,中国医学科学院北京协和医学院医药生物技术研究所抗感染药物研究北京市重点实验室(颜海燕、李玉环)
流行性感冒,又称流感,是由流感病毒(influenza A virus,IAV)引起的急性呼吸道疾病,在全球范围内具有较高的发病率和死亡率。流感病毒导致季节性大流行和人畜共患疾病的暴发,给全球经济带来严重的损失并对世界范围内的公共卫生产生威胁。疫苗和抗病毒药物是预防和治疗流感病毒的主要手段,但因流感病毒易发生抗原漂移和抗原转换,疫苗防治效率低。目前临床上的抗病毒药物主要有:M2 离子通道抑制剂金刚烷胺和金刚乙胺;神经氨酸酶抑制剂扎那米韦、拉尼米韦及其成酯后的辛酸拉尼米韦、磷酸奥司他韦和帕拉米韦;RNA 聚合酶抑制剂法匹拉韦和处于临床试验阶段的匹莫地韦;CAP 依赖性核酸内切酶抑制剂巴洛沙韦[1]。这些直接抗病毒药物的反复使用加剧流感耐药株的产生。因此为克服抗病毒药物耐药问题,开发靶向宿主的抗病毒药物成为防治流感病毒的新策略。靶向宿主的抗病毒药物主要分为两类:一是抑制参与流感病毒复制的宿主蛋白;二是增强流感病毒复制相关的宿主限制因子。本文对靶向流感病毒进入、复制和释放过程所需宿主蛋白、细胞因子和信号通路的抗流感病毒药物进行综述(图 1)。
1 靶向流感病毒进入阶段的宿主靶点及抗流感病毒抑制剂
流感通过病毒膜蛋白血凝素(heamagglutinin,HA)识别细胞表面的唾液酸受体附着在宿主细胞上,由宿主蛋白酶水解 HA,启动内吞作用进入宿主细胞。唾液酸是一个九碳单糖,家族中最常出现的成员是 N-乙酰神经氨酸,到目前为止已报道约 80 种唾液酸衍生物[2],主要位于细胞膜表面,在调节流感病毒的附着和进入中起着重要作用。DAS181(fludase)是一种宿主靶向的重组唾液酸酶融合蛋白,对多种 IAV 毒株具有良好的抑制作用[3]。其作用是除去呼吸道上皮细胞表面唾液酸,防止流感病毒附着,目前正处于流感治疗II期临床试验阶段[4]。
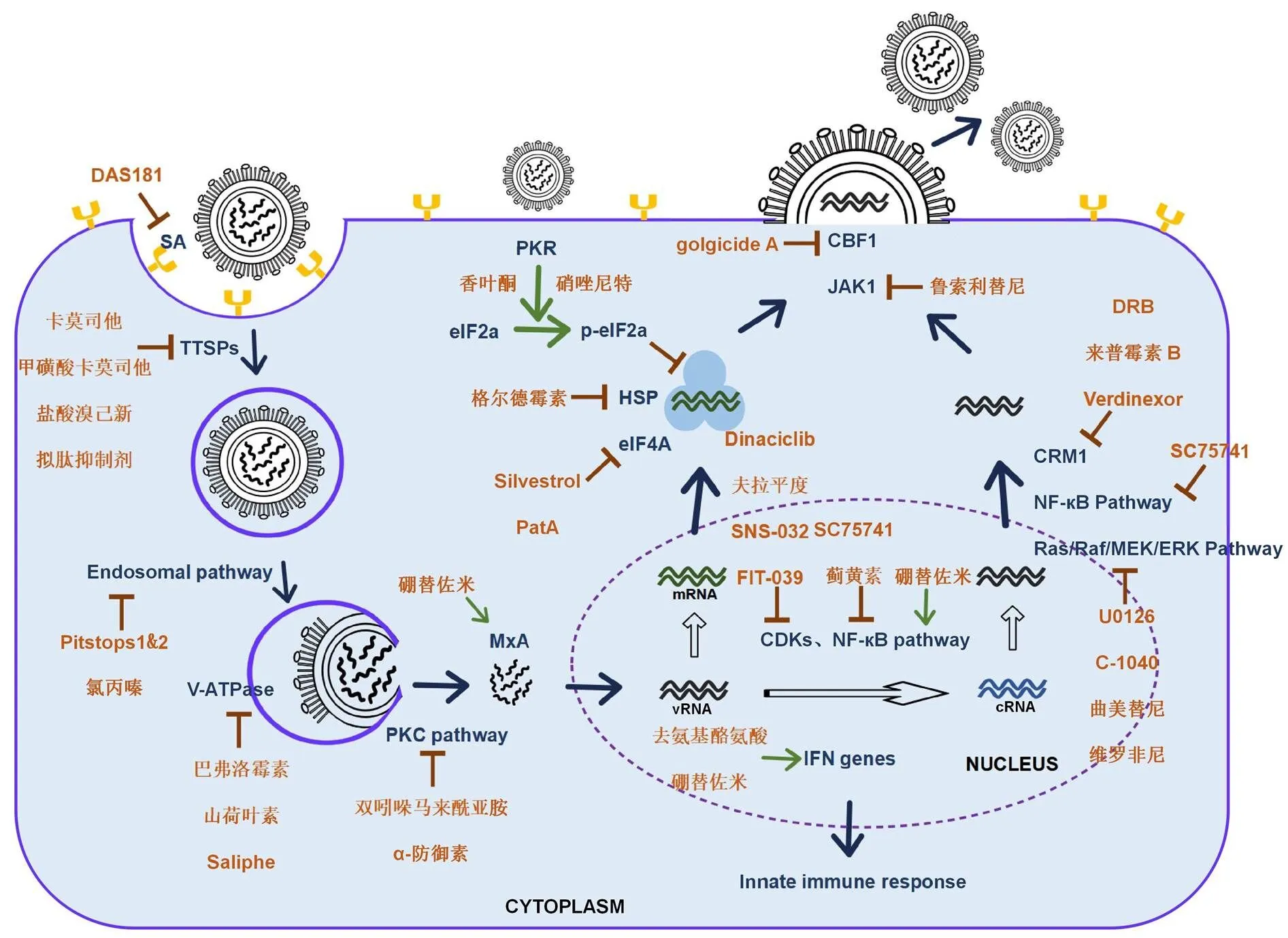
图 1 参与流感病毒复制的宿主靶点及抗病毒药物
II 型跨膜丝氨酸蛋白酶(type II transmembrane serine proteases,TTSPs)是一种主要水解 HA 的宿主蛋白酶[5]。TTSPs 家族包含跨膜蛋白酶丝氨酸 2(TMPRSS2)、跨膜蛋白酶丝氨酸 4(TMPRSS4)、人呼吸道胰蛋白酶样蛋白酶(HAT)等,实验数据表明这些宿主蛋白酶在流感病毒、冠状病毒和副黏病毒激活中起作用[6]。卡莫司他、甲磺酸卡莫司他、盐酸溴己新和拟肽抑制剂等以宿主蛋白酶为靶点抑制流感病毒感染[5-7]。因此以宿主蛋白酶为靶点,可开发广谱抗病毒药物。
内吞主要包括网格蛋白和胞膜窖依赖或非依赖性内吞途径及巨胞饮作用途径[8]。其中网格蛋白介导的内吞作用(clathrin-mediated endocytosis,CME)是主要途径。2011 年,von Kleist 等[9]鉴定并验证了网格蛋白末端结构域的小分子抑制剂 pitstops1 和 2,可抑制网格蛋白介导的内吞作用,进而阻断病毒入侵,并对细胞活力无明显影响。氯丙嗪作为网格蛋白依赖性内吞作用的抑制剂,最早被用于精神分裂症的治疗。Daniel 等[10]通过超分辨率显微镜检查发现,氯丙嗪既不阻断由衔接蛋白(AP-2)募集的网格蛋白,也不阻断 AP-2 的募集,而是作用在网格蛋白包被囊泡形成的下游,且有数据表明发动蛋白是它们在胞吞作用中可能的细胞内靶标。文献[11]报道,氯丙嗪与抑制细胞内吞抑制剂 EIPA 协同作用可以降低流感病毒感染 MDCK 细胞的能力。因此,可以通过开发抗流感病毒药物联合用药来抑制病毒进入宿主细胞。
位于细胞器和细胞质膜上的液泡型 ATP 酶(V-ATPase),通过泵送质子酸化晚期内体。当病毒处于酸性环境中,HA 发生构象改变,促进膜融合,形成融合孔释放 vRNPs 到细胞质中,完成病毒脱衣壳,因此可通过抑制 V-ATPase 活性干扰流感病毒感染。巴弗洛霉素属于大环内酯类抗生素家族。有文献报道,巴弗洛霉素可抑制 V-ATPase,降低内体酸化和溶酶体数量,从而抑制A 型和 B 型流感病毒复制[12-13]。山荷叶素是从分离得到的一种天然化合物,已被鉴定为一种新型 V-ATPase 抑制剂,用于治疗胃癌和抑制人破骨细胞内溶酶体酸化[14-15]。目前,Chen 等[16]发现山荷叶素在 MDCK 和 A549 细胞系中,对包括耐药病毒株在内的多种 IAV 毒株均有抑制作用。此外,联合使用山荷叶素和奥司他韦或山荷叶素和金刚烷胺治疗显示出增强的抗病毒效果和细胞保护作用。Saliphenylhalamide(Saliphe)作为一种抗癌药物,可以抑制 V-ATPase 活性,通过抑制内体的酸化,降低子代病毒的产生[17]。
蛋白激酶 C(protein kinase C,PKC)是一种丝氨酸/苏氨酸蛋白激酶(Ser/Thr protein kinase,STK),调节细胞增殖、分化、凋亡和血管生成等多个过程,在包膜病毒进入宿主细胞过程中发挥重要作用。双吲哚马来酰亚胺(GF109203X)是高度特异性 PKC 抑制剂,用其处理细胞,对大多数 PKC 亚型具有活性,在阻断流感病毒内体运输和可逆地抑制病毒颗粒脱壳进入细胞质这一过程发挥重要作用[18]。防御素是小型阳离子抗菌肽,分为两个主要亚家族:α-防御素(HNP-1)和 β-防御素。据报道,HNP-1 对多种病毒具有抑制活性,在感染后不久加入 HNP-1,可以有效抑制流感病毒复制和病毒蛋白合成,其抗病毒机制不是直接作用在病毒上,而是通过调节对病毒生命周期重要的 PKC 通路介导[19-20]。
2 靶向流感病毒复制阶段的宿主靶点及抗流感病毒抑制剂
当释放在胞质中的病毒核糖核蛋白(vRNPs)进入细胞核后,在细胞周期依赖性蛋白激酶(CDKs)和 RNA 聚合酶II(Pol II)等宿主因子的作用下完成病毒 RNA(vRNA)的转录。这些宿主蛋白成为治疗药物开发的靶点,以阻止病毒的转录,其中 CDKs 的化学抑制剂获得广泛关注。CDKs 属于 STK 家族,在调节细胞周期,病毒的基因复制和转录[21]等过程中发挥重要作用。Perwitasari 等[22]在通过高通量方法筛选出的 273 个对流感病毒 H7N9 毒株有抑制作用的化合物中,发现 CDK1/2/5/9 抑制剂 dinaciclib 和 CDK1/2/4/6 抑制剂夫拉平度对 A/California/04/09(H1N1)和 A/Philippines/2/82-X79(H3N2)流感病毒毒株有抑制作用。这两种药物是通过抑制宿主 Pol II活性导致 vRNA 转录水平降低来发挥作用,其中 dinaciclib 对流感病毒的抑制作用比夫拉平度高 12 倍。SNS-032(CDK2/7/9 抑制剂)具有抑制病毒基因表达的作用,与未经治疗的小鼠高达 80% 的死亡率相比,SNS-032 可以保护小鼠免受 H1N1 型流感病毒的感染[23]。然而,广谱 CDKs 抑制剂通常会对细胞周期进程产生负面影响,并引起不良反应,导致抗病毒药物的开发困难。Yamamoto 等[21]研发了CDK9 抑制剂 FIT-039,可以抑制单纯疱疹病毒(herpes simplex virus,HSV)、人巨细胞病毒(human cytomegalovirus,HCMV)、人腺病毒(human adenovirus,HAdV)等 DNA 病毒的复制,临床前研究未见明显的副作用,提示 FIT-039 是一种有潜力的化合物。综合以上结果表明,进一步开发 CDK 抑制剂在控制流感病毒感染方面具有广阔的前景。
流感病毒蛋白质合成完全依赖于宿主细胞的翻译机制,病毒 mRNA 通过核孔复合体转运出核,在核糖体上进行翻译,内质网上合成,并通过高尔基体转运至质膜。热休克蛋白(heat shock protein,HSP)和真核起始因子(eIF)等密切参与此过程,因此是重要的抑制流感病毒的宿主靶点。
热休克蛋白是一大类参与蛋白质折叠和合成的应激蛋白质,主要根据其分子量进行分类,包括 HSP27、HSP40、HSP60、HSP70、HSP90 和大型热休克蛋白。格尔德霉素于 1970 年从吸水链霉菌中分离出来,是第一个报道的 Hsp90 抑制剂,其与 Hsp90 的 N 末端三磷酸腺苷结合口袋结合,抑制蛋白伴侣的功能。Wang 等[24]在小鼠体内进行格尔德霉素抗 H5N1 流感病毒实验,发现格尔德霉素可以延长小鼠的存活时间,显著减少肺损伤、肺部炎症及降低病毒载量。因格尔德霉素水溶性差,不良反应明显,后续针对格尔德霉素进行结构改造修饰,合成了高水溶性、低毒性的 17-AAG、17-DMAG。Chase 等[25]对格尔德霉素及其衍生物 17-AAG 进行体外抗流感病毒实验,显示病毒滴度在感染早期降低1 ~ 2 log。因此,Hsp90 抑制剂可能是一类新型的抗流感病毒的化合物。
真核翻译起始因子 4A(eIF4A)是一种解旋酶,通过解开 mRNA 5' 非翻译区来促进翻译起始前复合体的组装。天然产物 pateamine A(PatA)和 silvestrol 的作用机制是抑制 eIF4A 功能并阻止蛋白质翻译和病毒基因组复制,从而使翻译抑制的 mRNA 在细胞质聚集形成一种高密度结构体,称为应激颗粒(stress granule,SGs)。据报道,silvestrol具有广谱抗病毒作用,可抑制 A549 和 MDCK 细胞中流感病毒的复制[26-27]。但 silvestrol 的抗病毒作用是完全可逆的,停止给药后可引起 SGs 快速溶解,并恢复病毒蛋白质合成[27];而 PatA 不可逆地结合 eIF4A,在停药后可长期维持流感病毒复制阻断和蛋白合成停滞[28]。这些研究表明,抑制 eIF4A 可以阻止病毒的翻译和复制,是一个有前景的抗病毒靶点。
双链 RNA 依赖性蛋白激酶 R(doublestranded RNA-dependent protein kinase R,PKR)是宿主抗病毒感染先天免疫的重要组成部分,由 dsRNA 激活,进一步激活下游底物 eIF2α,具有诱导病毒和细胞基因翻译关闭的广泛抗病毒活性,但流感病毒 NS1 蛋白 N 末端的 35 和 46 位氨基酸可抑制 PKR 激活[29-30]。eIF2α 是真核起始复合物的 315 个氨基酸亚单位,当 eIF2α 未磷酸化时,蛋白质翻译以正常速率发生,当 eIF2α 被 PKR 等激酶磷酸化时,导致 mRNA 翻译受阻,蛋白质合成下调,从而形成 SGs。据报道,抗溃疡药物香叶酮通过增强 PKR 基因的表达,促进 PKR 及 eIF2α 磷酸化,以对抗流感病毒感染[31-32]。硝唑尼特为一种广谱抗病毒活性的噻唑烷,在体外研究显示,硝唑尼特可抑制多种流感病毒毒株的复制,与奥司他韦或扎那米韦联用,具有协同抗病毒作用[33]。硝唑尼特抗病毒机制包括诱导细胞内 PKR 和 eIF2α 磷酸化;消耗细胞内 Ga+储备[34-35]。因此,可以诱导 eIF2α 磷酸化来开发广谱抗病毒药物。
流感病毒感染宿主细胞,至少被 Toll 样病毒受体(Toll-like receptors,TLR)和视黄酸诱导基因(RIG)-1样受体(retinoic acid-inducible gene 1-like receptor,RLR)这两种病原体识别受体(pathogen recognition receptors,PRRs)识别,诱导 I 型干扰素(IFN)分泌,进而激活数百种干扰素刺激基因产物(IFN stimulated genes,ISG),激活适应性免疫系统。研究表明,ISG 具有广泛的抗病毒能力,可降解病毒核酸,抑制病毒基因表达,并作为 PRRs 扩增 IFN 信号[36-37]。例如,抗黏病毒蛋白(Mx)是 IFN 诱导的具有广泛抗病毒活性的关键细胞内限制因子,存在于几乎所有脊椎动物中。人体内 MxA 通过靶向 vRNPs 显示出有效抗流感病毒活性:一是将释放的 vRNPs 保留在细胞质中;二是阻断来自 cRNA 拷贝的 vRNA 二次转录[38]。微生物代谢物去氨基酪氨酸(desaminotyrosine,DAT)通过增强I型 IFN 信号和减少肺免疫病理损伤来阻断 IAV 感染[39]。
核因子-кB(nuclear factor-kappa B,NF-кB)通路是流感病毒感染后被 PRRs 激活的另一重要途径,在参与炎症反应,干扰流感病毒复制,抵抗病毒感染中发挥重要作用[40-42]。因此,NF-кB 通路是治疗流感的重要靶标。硼替佐米(PS-341),是一种临床批准的抗癌药物,其抗流感病毒活性不取决于可能的细胞毒性或促凋亡作用,而是诱导了 IкB 的降解和 NF-кB 以及 JNK/AP-1 途径的激活,诱导I 型 IFN 应答,使抗病毒基因(如IL-6、MxA)的表达增强,对I 型 IFN 缺乏的 Vero 细胞中的病毒滴度无影响[43]。蓟黄素是从植物茵陈蒿中提取的黄酮类化合物之一,能有效抑制甲型流感病毒株,包括 A/tianjinjinnan/15/2009(H1N1)和 A/JiangXi/312/2006(H3N2)。其作用机制主要是通过下调丝裂原活化蛋白激酶(MAPKs)(p38 和 JNK)信号通路的磷酸化,影响 NF-кB 信号通路的磷酸化,抑制炎症因子如 IL-1β、IL-8、IL-10 和 TNF-α 的水平而发挥抗流感病毒作用[44]。此外,SC75741 抑制 NF-кB 活化,介导 RNPs 出核,并在治疗小鼠免受流感病毒感染所需浓度方面没有显示出副作用,且阻碍耐药毒株的发展,是一种有前途的靶向宿主的候选药物[45-46]。
3 靶向流感病毒释放阶段的宿主靶点及抗流感病毒抑制剂
在流感病毒复制后期,利用宿主细胞蛋白运输系统将新合成的 vRNPs 和病毒结构蛋白转运至质膜区域装配成子代病毒,随后萌芽释放成熟病毒颗粒。
染色体维持蛋白 1(chromosome region maintenance 1,CRM1)是抗病毒药物开发新的潜在靶点,其介导流感病毒核输出功能[47]。二氯苯并咪唑呋喃型核糖苷(5,6-dichloro- 1-β-D-ribofuanosyl-H-benzimidazole,DRB)和来普霉素B(LMB)抑制 CRM1 和 TAP-P15 途径的核转运,完全阻断了流感病毒输出[48],但 LMB 在体内有毒性,不适宜治疗流感。Verdinexor(KPT-335)是一种新型口服小分子核输出选择性抑制剂(SINE),研究证明 verdinexor 在体内外均有效抑制流感病毒,其作用是阻断 CRM1 介导的 vRNPs 核输出和抑制 NF-кB 激活,从而减少细胞因子的产生并消除病毒相关的免疫病理学,同时具有最小毒性[49-50]。
ERK 通路主要介导 Ras/Raf/MEK/ERK 级联途径,被认为是流感病毒激活的标志信号通路,对新合成的 vRNPs 的核输出及细胞生长、分化和存活都具有重要作用。特异性MEK 抑制剂 U0126 可以显著降低流感病毒复制,经鼻给予 U0126 可以保护小鼠免受流感病毒的感染,且不会产生任何副作用[51]。CI-1040 是一种三磷酸腺苷非竞争性 MEK1/2 抑制剂,通过阻止流感病毒 RNP 复合物的形成,显著降低 A549 细胞中流感病毒滴度,而且对高致病性禽流感毒株、达菲耐药株等多种流感病毒毒株均有效。此外,CI-1040 可以减少流感病毒感染小鼠肺组织中病毒量达 80% 以上[52]。MEK1/2 抑制剂曲美替尼是第一个获得FDA 批准的用于治疗恶性黑色素瘤的药物[53-54],可干扰子代病毒 vRNPs 的输出,具有广泛的抗病毒活性,在体外和体内均可有效阻断不同流感病毒亚型的复制[55]。B-Raf(V600E)是人类癌症中常见的突变基因,B-Raf(V600E)抑制剂维罗非尼可以有效阻止 Raf/MEK/ERK 信号级联的激活,对多种不同 IAV 毒株具有广泛的抗病毒活性。维罗非尼通过抑制凋亡诱导细胞因子的表达,抑制病毒诱导的 A549 细胞凋亡,导致病毒蛋白表达受阻。此外,维罗非尼可以抑制 IAV 诱导的其他信号级联的激活,尤其是 MAPKs(p38 和 JNK)通路等。多靶点抗病毒模式不易产生耐药性,因此使用维罗非尼对抗流感病毒感染是一种非常有希望和创新的抗病毒干预方法[56]。
Watanabe 等[57]验证发现与流感病毒相互作用的宿主因子 GBF1 和 JAK1 是抗病毒药物的潜在靶点。Golgicide A 抑制 GBF1,该宿主因子与 M2 发生免疫共沉淀,并参与 HA 和 NA 在细胞内的转运;鲁索利替尼(INCB18424)抑制 JAK1,该宿主因子与几种病毒蛋白发生免疫共沉淀,抑制子代病毒颗粒形成。
4 展望
流感大流行和人畜共患的流感是严重的公共卫生问题,世界卫生组织统计全球每年导致 25 万~ 50 万人死亡。流感病毒变异快、易耐药,直接针对病毒靶点的药物难以成功对抗流感病毒的流行,迫切需要研发新的作用机制的药物。流感病毒在生命复制周期的每个阶段都依赖于宿主细胞,病毒与其宿主细胞之间的相互作用决定了感染的结果,包括子代病毒生产的效率、趋向性和致病性。因此靶向宿主成为抗病毒药物研发的热点,特别适用于像流感病毒这类病程短的急性病毒性疾病。鉴于此,本文总结了近年来针对流感病毒复制所需宿主蛋白和细胞信号通路的抗病毒药物。相信随着分子病毒学、各种组学研究等技术的进步加深对病毒-宿主相互作用的深入理解,有望扩大潜在抗流感病毒宿主靶点的数量及靶向宿主抗流感病毒药物的研发。
[1] Cai Y, Ding XL, Zhang JB, et al. Survey on anti-influenza drug synthesis. Chin J Chem Educ, 2019, 40(12):5-18. (in Chinese)
蔡岩, 丁晓丽, 章聚宝, 等. 抗流感病毒药物合成研究纵览. 化学教育, 2019, 40(12):5-18.
[2] Schauer R, Kamerling JP. Exploration of the sialic acid world. Adv Carbohydr Chem Biochem, 2018, 75:1-213.
[3] Malakhov MP, Aschenbrenner LM, Smee DF, et al. Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection. Antimicrob Agents Chemother, 2006, 50(4):1470-1479.
[4] Nicholls JM, Moss RB, Haslam SM. The use of sialidase therapy for respiratory viral infections. Antiviral Res, 2013, 98(3):401-409.
[5] Wu C, Wen ZY, Chen WY, et al. The cleavage of 2009 pandemic influenza HA protein by TMPRSS4 and induced cell fusion activity in vitro. Chin J Prev Vet Med, 2013, 35(7):517-520. (in Chinese)
吴超, 温志远, 陈伟业, 等. 跨膜丝氨酸蛋白酶-4对2009甲型H1N1流感病毒HA的切割及其融合活性研究. 中国预防兽医学报, 2013, 35(7):517-520.
[6] Laporte M, Naesens L. Airway proteases: an emerging drug target for influenza and other respiratory virus infections. Curr Opin Virol, 2017, 24:16-24.
[7] Li CC, Wang XJ, Wang HR. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov Today, 2019, 24(3):726-736.
[8] Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem, 2010, 79:803-833.
[9] von Kleist L, Stahlschmidt W, Bulut H, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell, 2011, 146(3):417-484.
[10] Daniel JA, Chau N, Abdel-Hamid MK, et al. Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic, 2015, 16(6):635-654.
[11] Rossman JS, Leser GP, Lamb RA. Filamentous influenza virus enters cells via macropinocytosis. J Virol, 2012, 86(20):10950-10960.
[12] Yeganeh B, Ghavami S, Kroeker AL, et al. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am J Physiol Lung Cell Mol Physiol, 2015, 308(3):L270-L286.
[13] Ochiai H, Sakai S, Hirabayashi T, et al. Inhibitory effect of bafilomycin A1, a specific inhibitor of vacuolar-type proton pump, on the growth of influenza A and B viruses in MDCK cells. Antiviral Res, 1995, 27(4):425-430.
[14] Sørensen MG, Henriksen K, Neutzsky-Wulff AV, et al. Diphyllin, a novel and naturally potent V-ATPase inhibitor, abrogates acidification of the osteoclastic resorption lacunae and bone resorption. J Bone Miner Res, 2007, 22(10):1640-1648.
[15] Shen W, Zou X, Chen M, et al. Effects of diphyllin as a novel V-ATPase inhibitor on gastric adenocarcinoma. Eur J Pharmacol, 2011, 667(1-3):330-338.
[16] Chen HW, Cheng JX, Liu MT, et al. Inhibitory and combinatorial effect of diphyllin, a v-ATPase blocker, on influenza viruses. Antiviral Res, 2013, 99(3):371-382.
[17] Denisova OV, Kakkola L, Feng L, et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza A virus infection. J Biological Chem, 2012, 287(42):35324-35332.
[18] Meineke R, Rimmelzwaan GF, Elbahesh H. Influenza virus infections and cellular kinases. Viruses, 2019, 11(2):171.
[19] Daher KA, Selsted ME, Lehrer RI. Direct inactivation of viruses by human granulocyte defensins. J Virology, 1986, 60(3):1068-1074.
[20] Salvatore M, García-Sastre A, Ruchala P, et al. α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J Infect Dis, 2007, 196(6):835-843.
[21] Yamamoto M, Onogi H, Kii I, et al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses. J Clin Invest, 2014, 124(8):3479- 3488.
[22] Perwitasari O, Yan X, O'Donnell J, et al. Repurposing kinase inhibitors as antiviral agents to control influenza A virus replication. Assay Drug Dev Technol, 2015, 13(10):638-649.
[23] Söderholm S, Anastasina M, Islam MM, et al. Immuno-modulating properties of saliphenylhalamide, SNS-032, obatoclax, and gemcitabine. Antiviral Res, 2016, 126:69-80.
[24] Wang C, Liu P, Luo J, et al. Geldanamycin reduces acute respiratory distress syndrome and promotes the survival of mice infected with the highly virulent H5N1 influenza virus. Front Cell Infect Microbiol, 2017, 7:267.
[25] Chase G, Deng T, Fodor E, et al. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology, 2008, 377(2):431-439.
[26] Müller C, Schulte FW, Lange-Grünweller K, et al. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antiviral Res, 2018, 150:123-129.
[27] Slaine PD, Kleer M, Smith NK, et al. Stress granule-inducing eukaryotic translation initiation factor 4A inhibitors block influenza A virus replication. Viruses, 2017, 9(12):388.
[28] Low WK, Dang Y, Schneider-Poetsch T, et al. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell, 2006, 20(5):709-722.
[29] Balachandran S, Roberts PC, Brown LE. et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity, 2000, 13(1):129-141.
[30] Schierhorn KL, Jolmes F, Bespalowa J, et al. Influenza A virus virulence depends on two amino acids in the N-terminal domain of its NS1 protein to facilitate inhibition of the RNA-dependent protein kinase PKR. J Virol, 2017, 91(10):e00198-17.
[31] Unoshima M, Iwasaka H, Eto J, et al. Antiviral effects of geranylgeranylacetone: enhancement of MxA expression and phosphorylation of PKR during influenza virus infection. Antimicrob Agents Chemother, 2003, 47(9):2914-2921.
[32] Ding L, Zhu T, Song Q, et al. HPLC-APCI-MS for the determination of teprenone in human plasma: method and clinical application.J Pharm Biomed Anal, 2007, 44(3):779-785.
[33] Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res, 2014, 110:94-103.
[34] Elazar M, Liu M, Mckenna SA, et al. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology, 2009, 137(5):1827-1835.
[35] Ashiru O, Howe JD, Butters TD. Nitazoxanide, an antiviral thiazolide, depletes ATP-sensitive intracellular Ca(2+) stores. Virology, 2014, 462-463:135-148.
[36] Li CC, Wang XJ, Wang HR. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov Today, 2019, 24(3):726-736.
[37] Bai SY, Yang Q, Qiu HJ. Antiviral mechanisms of interferon-stimulated genes. Acta Microbiol Sinica, 2018, 58(3):361-371. (in Chinese)
白思宇, 杨倩, 仇华吉. 干扰素刺激基因的抗病毒机制. 微生物学报, 2018, 58(3):361-371.
[38] Villalón-Letelier F, Brooks AG, Saunders PM, et al. Host cell restriction factors that limit influenza A infection. Viruses, 2017, 9(12):E376.
[39] Steed AL, Christophi GP, Kaiko GE, et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science, 2017, 357(6350):498-502.
[40] Ludwig S, Planz O. Influenza viruses and the NF-kappaB signaling pathway - towards a novel concept of antiviral therapy. Biol Chem, 2008, 389(10):1307-1312.
[41] Pasparakis M. Role of NF-κB in epithelial biology. Immunol Rev, 2012, 246(1):346-358.
[42] Lee SM, Yen HL. Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res, 2012, 96(3):391-404.
[43] Dudek SE, Luig C, Pauli EK, et al. The clinically approved proteasome inhibitor PS-341 efficiently blocks influenza A virus and vesicular stomatitis virus propagation by establishing an antiviral state. J Virol, 2010, 84(18):9439-9451.
[44] Yan H, Wang H, Ma L, et al. Cirsimaritin inhibits influenza A virus replication by downregulating the NF-κB signal transduction pathway. Virol J, 2018, 15(1):88.
[45] Haasbach E, Reiling SJ, Ehrhardt C, et al. The NF-kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza A virus. Antiviral Res, 2013, 99(3):336-344.
[46] Ehrhardt C, Rückle A, Hrincius ER, et al. The NF-κB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol, 2013, 15(7):1198-1211.
[47] Chutiwitoonchai N, Mano T, Kakisaka M, et al. Inhibition of CRM1-mediated nuclear export of influenza A nucleoprotein and nuclear export protein as a novel target for antiviral drug development. Virology, 2017, 507:32-39.
[48] Yadav V, Panganiban AT, Honer Zu Bentrup K, et al. Influenza infection modulates vesicular trafficking and induces Golgi complex disruption. VirusDisease, 2016, 27(4):357-368.
[49] Pickens JA, Tripp RA. Verdinexor targeting of CRM1 is a promising therapeutic approach against RSV and influenza viruses. Viruses, 2018, 10(1):E48.
[50] Perwitasari O, Johnson S, Yan X, et al. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza A virus replication in vitro and in vivo. J Virol, 2014, 88(17):10228-10243.
[51] Droebner K, Pleschka S, Ludwig S, et al. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res, 2011, 92(2): 195-203.
[52] Haasbach E, Müller C, Ehrhardt C, et al. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antiviral Res, 2017, 142:178-184.
[53] Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Canc Res, 2011, 17(5):989-1000.
[54] Wright CJ, Mccormack PL. Trametinib: first global approval. Drugs, 2013, 73(11):1245-1254.
[55] Schräder T, Dudek SE, Schreiber A. et al. The clinically approved MEK inhibitor Trametinib efficiently blocks influenza A virus propagation and cytokine expression. Antiviral Res, 2018, 157:80-92.
[56] Holzberg M, Boergeling Y, Schräder T, et al. Vemurafenib limits influenza A virus propagation by targeting multiple signaling pathways. Front Microbiol, 2017, 8:2426.
[57] Watanabe T, Kawakami E, Shoemaker JE, et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe, 2014, 16(6):795-805.
“重大新药创制”国家科技重大专项(2018ZX09711003);中国医学科学院医学与健康科技创新工程(2017-I2M-3-010);佳木斯大学优秀学科团队项目(JDXKDT-2019005)
于莲,Email:jdyulian@163.com
2019-11-28
10.3969/j.issn.1673-713X.2020.03.015
