HPLC法测定对乙酰氨基酚片中有关物质的含量
2020-06-09杨莉梅勇龙涛罗磊陈小雪
杨莉 梅勇 龙涛 罗磊 陈小雪

中圖分类号 R917 文献标志码 A 文章编号 1001-0408(2020)10-1233-06
DOI 10.6039/j.issn.1001-0408.2020.10.15
摘 要 目的:建立测定对乙酰氨基酚片中有关物质含量的方法。方法:采用高效液相色谱法。色谱柱为Agilent 5HC-C8,流动相A、B分别为甲醇-水-冰醋酸(50 ∶ 950 ∶ 1,V/V/V)和甲醇-水-冰醋酸(500 ∶ 500 ∶ 1,V/V/V)(梯度洗脱),流速为0.9 mL/min,检测波长为254 nm,柱温为40 ℃,进样量为5 μL。结果:该色谱条件下,对乙酰氨基酚片中的主药(对乙酰氨基酚)、6个已知杂质(对氨基酚、对氯苯乙酰胺和杂质A、B、D、F)、3个制剂特定辅料(羟苯甲酯、羟苯乙酯和羟苯丙酯)和1个未知杂质的分离度均大于1.5。6个已知杂质检测质量浓度的线性范围分别为0.539~1.617、0.026~0.384、0.237~17.799、0.257~19.271、0.239~17.955、0.246~18.462 μg/mL(r≥0.999 8),杂质A、B、D、F的校正因子分别为2.9、1.0、1.2、6.2;检测限分别为0.009 6、0.024 2、0.164 0、0.051 1、0.055 9、0.422 0 ng,定量限分别为0.032 0、0.080 6、0.546 0、0.170 0、0.186 0、1.406 0 ng;平均回收率为95.96%~111.09%(RSD为0.05%~2.42%);精密度试验的RSD均小于15%,且耐用性良好。3批样品均检出了对氨基酚(均为0.006%)、杂质B(0.016%~0.017%)、未知杂质(0.002 0~0.002 1%),未检出对氯苯乙酰胺和杂质A、D、F。结论:该方法专属性强、准确度高,可用于对乙酰氨基酚片有关物质的测定。
关键词 高效液相色谱法;对乙酰氨基酚片;有关物质;含量
Content Determination of Related Substances in Paracetamol Tablets by HPLC
YANG Li,MEI Yong,LONG Tao,LUO Lei,CHEN Xiaoxue(Chongqing Cathay Corning Pharmaceutical Co.,Ltd., Chongqing 401520,China)
ABSTRACT OBJECTIVE: To establish the method for content determination of related substances in Paracetamol tablets. METHODS:HPLC method was adopted. The determination was performed on Agilent 5HC-C8 column with mobile phase A consisted of methanol-water-glacial acetic acid (50 ∶ 950 ∶ 1, V/V/V) and mobile phase B consisted of methanol-water-glacial acetic acid (500 ∶ 500 ∶ 1, V/V/V) (gradient elution) at the flow rate of 0.9 mL/min. The detection wavelength was set at 254 nm, and column temperature was 40 ℃. The sample size was 5 μL. RESULTS: Under the chromatographic condition, the resolutions of main component (paracetamol), 6 known impurities (p-aminophenol, p-chloroacetanilide, impurity A, B, D, F), 3 specific excipients (methyl hydroxybenzoate, ethyl hydroxybenzoate, propyl hydroxybenzoate) and 1 unknown impurity were all higher than 1.5. The linear range of 6 known impurities were 0.539-1.617, 0.026-0.384, 0.237-17.799, 0.257-19.271, 0.239-17.955, 0.246-18.462 μg/mL(r≥0.999 8), respectively. Correction factors of impurity A, B, D, F were 2.9, 1.0, 1.2, 6.2. The limits of detection were 0.009 6, 0.024 2, 0.164 0, 0.051 1, 0.055 9, 0.422 0 ng; the limits of quantitation were 0.032 0, 0.080 6, 0.546 0, 0.170 0, 0.186 0, 1.406 0 ng. Average recoveries were 95.96%-111.09%(RSDs were 0.05%-2.42%). The RSDs precision test were low than 15%, and the durability were good. p-aminophenol (all were 0.006%), impurity B (0.016%-0.017%) and unknown impurity (0.002 0%-0.002 1%) were detected in 3 batches of sample. p-choroacetanilide, impurity A, D and F were not detected. CONCLUSIONS: The method is specific, accurate and suitable for the determination of related substance in Paracetamol tablets.
KEYWORDS HPLC; Paracetamol tablets; Related substance; Content
对乙酰氨基酚片为乙酰苯胺类解热镇痛药,临床上主要用于缓解轻中度疼痛,如能最小限度地缓解膝关节的疼痛[1],是轻型骨关节炎短期镇痛的首选药物[2]。作为常用的解热镇痛药,对乙酰氨基酚片在治疗剂量内安全、可靠;但当剂量过大时,则可能导致急性肝功能衰竭[3],同时还可能引起血压上升[4]、哮喘[5]、肝毒性[6]及胃肠道反应等不良反应[7-8],因此使用时应严格遵循其使用原则。《国家药品安全“十二五”规划》[9]指出,对乙酰氨基酚片为一致性评价品种,参照处方信息,原研药由对乙酰氨基酚、预胶化淀粉、二氧化硅、海藻酸、聚维酮K30、羟苯甲酯、羟苯乙酯和羟苯丙酯等组成,为有刻痕的胶囊形片剂[10-11]。该产品中可能存在的主要杂质为对氨基酚、对氯苯乙酰胺和杂质A、B、D、F等,这些杂质大多含有潜在基因毒性片断;但除对氨基酚外,2015年版《中国药典》(二部)均未对上述其余杂质进行控制[12]。此外,为避免因生产运行中频繁清洗设备所造成的污染,处方中加入的羟苯甲酯、羟苯乙酯和羟苯丙酯[10]均可在分析时被检出,作为制剂特定辅料,上述3种成分在杂质含量计算时应予以扣除。鉴于现行国家药品标准[12]中并无本品有关物质检测项,且文献报道的方法[13-14]均不能将已知杂质、未知杂质与主成分完全分离,本研究建立了测定对乙酰氨基酚片中有关物质含量的方法,以期能同时分离已知杂质、未知杂质、主成分及片剂中加入的特定辅料,为产品质量的保证以及对乙酰氨基酚片一致性评价中的杂质研究提供方法支持。
1 材料
1.1 仪器
LC-2030型高效液相色谱仪,包括在线脱气机、二元高压泵、自动进样器、二极管阵列检测器(日本Shimadzu公司);MS205DU型电子天平(瑞士Mettler Toledo公司);KQ5200B型超声波清洗器(昆山市超声仪器有限公司)。
1.2 药品与试剂
对乙酰氨基酚片(重庆国泰康宁制药有限责任公司,批号:180501、180502、180503,规格:0.5 g);对乙酰氨基酚对照品(批号:100018-201610,纯度:99.9%)、对氨基酚对照品(批号:100802-201203,纯度:100%)、对氯苯乙酰胺对照品(批号:100850-201803,纯度:100%)均购自中国食品药品检定研究院;杂质A对照品(批号:36404,纯度:99.8%)、杂质B对照品(批号:43849,纯度:99.9%)、杂质D对照品(批号:76103,纯度:100%)、杂质F对照品(批号:66550,纯度:99.9%)均购自英国LGC公司;甲醇为色谱纯,冰醋酸等其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 溶液的制备
2.1.1 杂质对照品溶液 取对氨基酚、对氯苯乙酰胺和已知杂质A、B、D、F对照品各适量,精密称定,用甲醇溶解并稀释,制成含对氨基酚1 mg/mL,对氯苯乙酰氨2.5 μg/mL,杂质A、B、D、F 800 μg/mL的混合溶液,作为杂质对照品贮备液;取上述对照品贮备液5 mL,用甲醇稀释至50 mL,作为杂质对照品溶液。
2.1.2 对照品溶液 取对乙酰氨基酚对照品适量,精密称定,用甲醇溶解并稀释,制成含对乙酰氨基酚20 μg/mL的溶液,作为对照品溶液。
2.1.3 供试品溶液 取对乙酰氨基酚片,研细,取细粉适量,精密称定,置于25 mL量瓶中,用甲醇溶解并稀释,制成含对乙酰氨基酚20 μg/mL的溶液,滤过,取滤液作为供试品溶液。
2.1.4 对照溶液 取“2.1.3”项下供试品溶液1 mL,置于100 mL量瓶中,用甲醇稀释至刻度,作为对照溶液。
2.1.5 对照品+杂质混合溶液 取“2.1.1”项下杂质对照品贮备液1 mL,置于10 mL量瓶中,用“2.1.2”项下对照品溶液稀释至刻度,摇匀,滤过,作为对照品混合溶液。
2.1.6 供试品+杂质混合溶液 取“2.1.1”项下杂质对照品贮备液1 mL,置于10 mL量瓶中,用“2.1.3”项下供试品溶液稀释至刻度,摇匀,滤过,取滤液,即得。
2.1.7 阴性溶液 以甲醇作为阴性溶液。
2.2 色谱条件
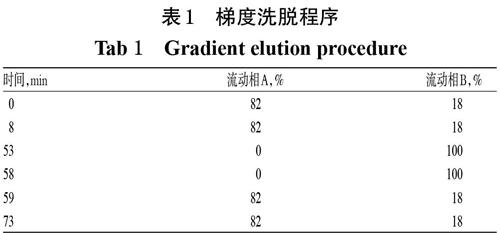
色谱柱:Agilent 5HC-C8(250 mm×4.6 mm,5 μm);流动相A:甲醇-水-冰醋酸(50 ∶ 950 ∶ 1,V/V/V),流动相B:甲醇-水-冰醋酸(500 ∶ 500 ∶ 1,V/V/V),梯度洗脱(程序见表1);流速:0.9 mL/min;柱温:40 ℃;检测波长:254 nm;进样量:5 μL。
2.3 系統适用性试验
取“2.1”项下阴性溶液、对照品+杂质混合溶液、供试品(批号:180501)溶液、供试品(批号:180501)+杂质混合溶液各5 μL,按“2.2”项下色谱条件进样测定,记录色谱图,详见图1。
由图1可见,在该色谱条件下,分别检出6个已知杂质(对氨基酚、对氯苯乙酰胺和杂质A、B、D、F)、1个未知杂质和3个制剂特定辅料(羟苯甲酯、羟苯乙酯和羟苯丙酯),各杂质与主成分(对乙酰氨基酚)的分离度以及各杂质间的分离度均大于1.5,表明该色谱条件专属性良好。
2.4 破坏试验
取对乙酰氨基酚片(批号:180501),研细,取细粉约33 mg,精密称定,置于25 mL量瓶中,加甲醇5 mL超声(功率:300 W,频率:40 kHz)处理5 min后,再分别置于高温(95 ℃水浴8 h)、酸(加1 mol/L盐酸溶液4 mL,室温放置21 h)、碱(加1 mol/L 氢氧化钠溶液4 mL,室温放置21 h)、光照(5 000 lx照射5 d)、氧化(加30%过氧化氢溶液4 mL,室温放置24 h)等条件下进行强制破坏,破坏结束后取出溶液,冷却至室温,用甲醇稀释至刻度(酸碱破坏样品需先用碱或酸调节pH为中性),滤过,取滤液按“2.2”项下色谱条件进样测定,记录色谱图,并以供试品溶液作为未破坏溶液进行对比,详见图2。结果,在上述破坏条件下,主成分与各已知杂质、未知杂质均能较好分离,表明上述条件对主成分和杂质的测定无影响。
2.5 线性关系和校正因子考察
取对乙酰氨基酚对照品适量,精密称定,加甲醇稀释,制成每1 mL约含0.1、5、12.5、20、25、37 μg的溶液,作为对乙酰氨基酚线性溶液;取对氨基酚对照品适量,精密称定,加甲醇稀释,制成每1 mL约含0.5、0.6、0.9、1.1、1.6 μg的溶液,作为对氨基酚线性溶液;取对氯苯乙酰胺对照品适量,精密称定,加甲醇稀释,制成每1 mL约含0.02、0.12、0.20、0.25、0.38 μg的溶液,作为对氯苯乙酰胺线性溶液;分别取杂质A、B、D、F对照品适量,精密称定,加甲醇稀释,制成每1 mL约含0.25、1.25、6.25、10.00、12.50、18.75 μg的溶液,作为单一杂质A、B、D、F线性溶液。取上述溶液按“2.2”项下色谱条件进样测定,记录峰面积。以峰面积为纵坐标(y)、进样质量浓度为横坐标(x)进行线性回归,并按以下公式计算校正因子,校正因子=对乙酰氨基酚线性方程的斜率/杂质线性方程的斜率。线性关系和校正因子考察结果详见表2。
注:“/”表示勿需计算校正因子,采用外标法计算杂质含量
Note:“/” means the correction factor dont need to be calculated,and the impurity content is calculated by external standard method
2.6 检测限和定量限考察
取“2.5”项下各杂质线性溶液中的最低质量浓度溶液,用甲醇继续逐级稀释,按“2.2”项下色谱条件进样测定,以能检测、定量的最低质量作为检测限和定量限,结果见表3、表4。
由表3和表4可见,各杂质检测量远低于各杂质的控制限度要求,各杂质能准确检测的量也远低于各杂质的控制限度要求,表明本方法能满足杂质检测灵敏度的要求。
2.7 精密度试验
(1)重复性:取“2.1”项下供试品溶液(批号:180501)适量,各6份,按“2.2”项下色谱条件进样测定,记录峰面积,采用外标法计算对氨基酚和对氯苯乙酰胺的含量;采用不加校正因子的主成分自身对照法计算杂质B和未知杂质的含量,采用加校正因子的主成分自身对照法计算杂质A、D、F的含量,结果见表5。
(2)中间精密度:分别由2名实验人员于不同时间按“2.1”项下方法制备供试品溶液(批号:180501),各6份,按“2.2”项下色谱条件进样测定,记录峰面积,按“2.7(1)”项下方法计算各杂质的含量,结果见表6。
由表5和表6可见,对氨基酚、杂质A、杂质B、杂质D、杂质F、未知杂质和总杂质含量的RSD均小于4%;对氯苯乙酰胺因含量极低,故RSD值偏大,但亦小于15%,表明本方法精密度良好。
2.8 加样回收率试验
取已知含量的对乙酰氨基酚片(批号:180501),共9份,精密称取适量,用甲醇制备成质量浓度为25 μg/mL的溶液,作为本底溶液;再分别加入杂质限度50%、100%、150%的对氨基酚对照品或对应杂质限度80%、100%、120%的杂质A、杂质B、杂质D、杂质F和对氯苯乙酰胺对照品,每质量浓度各3份,按“2.2”项下色谱条件进样测定,记录峰面积,计算加样回收率。结果,对氨基酚、杂质A、杂质B、杂质D、杂质F和对氯苯乙酰胺的平均加样回收率为96.89%~99.53%,RSD均小于2.0%,表明本方法准确度良好,详见表7。
2.9 稳定性试验
取“2.1.3”项下供试品溶液适量(批号:180501),分别置于室温和冰箱(温度1~4 ℃)中,于0、2、4、6、8、12 h时取样,按“2.2”项下色谱条件进样测定,记录峰面积,按“2.7(1)”项下方法计算各杂质的含量。结果,室温放置的供试品溶液中,对氨基酚峰面积随时间的延长而明显增加,含量的RSD大于2%(n=6);杂质B含量的RSD为1.73%(n=6),未知杂质含量的RSD为0.75%(n=6),其余已知杂质在各时间点均未检出,但溶液颜色由无色变成了黄色。由此可见,供试品溶液不宜在室温下放置,应临用新制。冰箱放置的供试品溶液中,其各已知杂质和未知杂质的峰面积在12 h内均无明显变化,RSD均小于2%(n=6),表明供试品溶液在1~4 ℃放置12 h内稳定。
2.10 耐用性试验
(1)改变色谱柱:取供試品溶液(批号:180501)适量,按“2.2”项下条件以不同色谱柱Agilent 5HC-C8(250 mm×4.6 mm,5 μm,色谱柱SN号分别为546817和541118)和Agela Venusil ASB C8(250 mm×4.6 mm,5 μm)进样测定,每个色谱柱测定9次,记录峰面积,并按“2.7(1)”项下方法计算含量,结果见表8。
(2)改变流速:取供试品溶液(批号:180501)适量,按“2.2”项下条件以不同流速(0.8、0.9、1.0 mL/min)进样测定,每种流速测定9次,记录峰面积,并按“2.7(1)”项下方法计算含量,结果见表8。
(3)改变柱温:取供试品溶液(批号:180501)适量,按“2.2”项下条件以不同柱温(30、40、45 ℃)进样测定,每个柱温测定9次,记录峰面积,并按“2.7(1)”项下方法计算含量,结果见表8。
由表8可见,在色谱条件轻微变动情况下,对乙酰氨基酚片中检出对氨基酚含量变化绝对值为0.002%,杂质B含量变化绝对值为0.001%,未知杂质含量变化绝对值为0.000 3%,总杂质含量变化绝对值为0.001 9%,其余已知杂质均未检出。这表明本方法在一定色谱柱变动、流速变动、柱温变动情况下耐用性良好。
2.11 样品中有关物质的测定
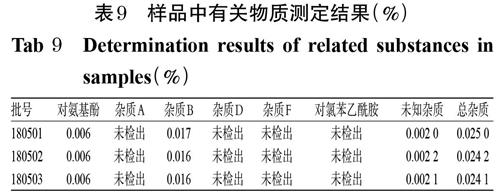
取3批样品各适量,按“2.1”项下方法制备成供试品溶液,按“2.2”项下色谱条件进样测定,记录峰面积,并按“2.7(1)”项下方法计算杂质含量,结果见表9。
由表9可见,3批对乙酰氨基酚片中均检出了对氨基酚、杂质B和未知杂质,均未检出杂质A、D、F和对氯苯乙酰胺。
3 讨论
本研究所建方法拟检测的对乙酰氨基酚片中的杂质A、D和对氯苯乙酰胺结构中含有苯乙酰胺结构片断,杂质B中含有苯丙酰胺结构片断,杂质F中含有硝基苯结构片断,对氨基酚中含有苯胺结构片断,上述结构片断均具有潜在的基因毒性。因此,有必要对这些杂质的含量进行检测和控制,以确保产品的安全性。但在2015年版《中国药典》(二部)中,除对对乙酰氨基酚片中对氨基酚的含量有所规定外,其他杂质含量均未作规定,无法确保产品的安全性,本研究可弥补其不足。
通过破坏试验可以看出,该色谱条件下,对于杂质A、B、D、F、对氨基酚和对氯苯乙酰胺之外的其余杂质也能得到有效检测,也就是说,在原料合成过程中可能产生的其他杂质(由EP 9.4对乙酰氨基酚质量标准[15]可知,还包括杂质C、E、H、L、M、N等)也可采用本方法有效检出。
综上所述,本研究所建方法简便易行,灵敏度高,准确度、稳定性、重复性、耐用性均较好,各已知杂质、未知杂质均能与主成分和特定辅料有效分离,可用于对乙酰氨基酚片的质量控制,有助于确保产品的安全有效和质量可控。
参考文献
[ 1 ] 赵元.对乙酰氨基酚:能最小限度地缓解膝关节炎的疼痛[J].心血管病防治知识,2017(5):52.
[ 2 ] 肖壮,唐涛.骨关节炎治疗药物的研究进展[J].中国药房,2016,27(35):5037-5040.
[ 3 ] 陳熠媛,陈硕崴,张谢,等.对乙酰氨基酚肝毒性研究进展[J].中国药房,2019,30(8):852-853.
[ 4 ] 洪思思.对乙酰氨基酚看起来对心脏是安全的[J].心血管病防治知识,2015(8):50.
[ 5 ] 伍灿,刘代顺.对乙酰氨基酚与哮喘发病的相关性研究进展[J].实用医学杂志,2015,31(5):854-855.
[ 6 ] 管斌.科学认识感冒药中“对乙酰氨基酚”的肝毒性[J].肝博士,2016(5):58-59.
[ 7 ] 丁召兴,成文娜,高菲,等.含对乙酰氨基酚药物的不良反应与合理使用[J].药学研究,2016(12):737-741.
[ 8 ] 蔡林,潘楠楠,石姗平.《国家基本药物目录》(2009版)中非甾体抗炎药物的临床安全性[J].中国药房,2013,24(12):1140-1142.
[ 9 ] 国务院.国家药品安全“十二五”规划[Z].2012-01-20.
[10] H·H·阿鲁尔.快速释放的对乙酰氨基酚片,中国:101466359A[P]. 2009-06-24.
[11] 刘元江,缪经纬,杨亚勇,等.不同厂家刻痕片及非刻痕片分剂量评价[J].中国药房,2012,23(29):2713-2715.
[12] 国家药典委员会.中华人民共和国药典:二部[S]. 2015年版.北京:中国医药科技出版社,2015:318-320.
[13] 余俊玲,钟瑜,徐伟斌,等. HPLC梯度洗脱法测定对乙酰氨基酚口服混悬液中有关物质[J].北方药学,2016,13(10):5-7.
[14] 郭波,唐秀玲,石磊,等.液相色谱-串联质谱法同时测定对乙酰氨基酚及其相关代谢产物[J].中国药师,2017,20(4):597-602.
[15] European Pharmacopoeia Supplemeat. European Pharmacopoeia:9.4[S]. 2017 edition. Strasbourg:the European Directorate for the Quality of Medicines & HealthCare of the Council of Europe,2017:5429-5430.
(收稿日期:2019-12-23 修回日期:2020-03-04)
(编辑:邹丽娟)
