HIST1H1E 基因突变致Rahman 综合征1 例并文献复习
2020-06-06罗燕飞孙光辉热衣兰木包尔汉迪丽胡麻居来提米热古丽买买提
李 燕 罗燕飞 孙光辉 热衣兰木·包尔汉 迪丽胡麻·居来提 米热古丽·买买提
新疆医科大学第一附属医院(新疆乌鲁木齐 830011)
Rahman 综合征(Rahman syndrome,RMNS,OMIM 617537)是一种罕见的常染色体显性遗传病,以轻度至重度智力障碍、发育落后和不同程度的躯体过度生长为特征。HIST1H1E基因(OMIM 142220)突变时,其C 末端结构域编码区发生移码和提前终止,产生的截短蛋白与DNA 的结合能力下降,并阻碍蛋白质之间的相互作用,从而影响人类表观遗传调控过程。目前国内尚无RMNS相关报道,本文将回顾分析1 例RMNS 患儿的临床资料及基因检测结果,并结合相关文献复习,总结其临床表型、遗传学特征以及诊断和治疗要点。
1 临床资料
患儿,男,2岁8个月,维吾尔族,以语言、运动发育落后为主诉就诊。患儿G2P1,足月顺产,出生体质量3 200 g。出生史、喂养史无异常。患儿自幼发育迟缓,4月龄抬头,9月龄独坐,18月龄独走,24月龄会说简单词语,现不能说完整句子,能理解他人说话。父母非近亲结婚,无家族性遗传病史。体格检查:生命体征平稳,身高98 cm(+0.95 SD),体质量19 kg。面容特殊:头围54 cm,发际线高,前额宽大,脸颊丰满,眦距宽,眼睑裂狭窄,牙列不齐,龋齿数个;心肺腹无异常;阴茎长2.5 cm,阴囊发育可,未触及睾丸,双侧腹股沟区可触及质韧包块;四肢无畸形,神经系统检查无异常。
因患儿存在发育落后,智力障碍,当地医院诊断不明确,建议上级医院进一步诊治;门诊考虑患儿遗传代谢病可能性大,同时患儿存在隐睾,建议完善手术,为全面评估患儿病情及制定诊疗计划,患儿家长要求住院明确诊断并行手术治疗。入院后实验室检查:常规血液学、生化、GnRH 激发试验、甲状腺功能、血同型半胱氨酸等均无异常;睾酮10.73 nmol/L;黄体生成素(LH)2.09 U/L,促卵泡生成素(FSH)6.03 U/L,LH/FSH 峰值0.43。因患儿存在智力障碍,语言运动发育落后等神经症状,为进一步排除代谢障碍性疾病,完善血氨基酸及肉碱谱检验,结果提示羟异戊酰肉碱增高,乙酰肉碱增高,鸟氨酸降低;尿有机酸谱检验结果未见典型有机酸代谢病改变。阴囊超声示双侧腹股沟区靠近阴囊根部可探及睾丸回声,右侧睾丸大小1.9 cm×0.6 cm,左侧睾丸大小1.8 cm×0.6 cm,回声均匀,鞘膜腔内未见积液。腕骨X光片示骨龄增大,符合年龄5.5岁(图1)。脊柱X光片示侧弯。超声心动图及垂体磁共振成像(MRI)未见明显异常。儿童发育筛查测验(dvelopmental screening test,DST):DQ<50、MI <48,智力符合年龄1岁3个月,运动符合年龄1岁9个月,社会适应符合年龄1岁6个月。

图1 患儿骨龄片
因患儿存在语言、运动发育落后及躯体过度生长,临床表现与典型的过度生长综合征。Sotos综合征表型相似,初步诊断Sotos综合征。为进一步明确诊断,经新疆医科大学伦理委员会审批,患儿父母签署书面知情同意书后,抽取患儿及其父母外周血各2 mL 进行全外显子组基因测序分析。利用Agilent SureSelect方法外显子捕获(Exome V6),llumina测序平台进行高通量测序,测序数据经NextGENe软件匹配分析后,用Ingenuity在线软件系统进行变异过筛及解释,候选变异经Sanger测序验证。结果显示,患儿HIST1H1E基因(NM_005321.2)存在碱基重复c.446 dupA,p.Ser 150 Glufs*46(杂合变异)[Chr 6(GRCh 37):g.26157064dup],见图2。该变异尚未在人类基因突变数据库(Human Gene Mutation Database,HGMD) 及gnomAD等数据库中收录(gnomAD∶dup=0.00)。患儿父母该位点均为正常基因型,提示该变异为新生突变(de novo)。经Alamut软件预测,此突变可以使氨基酸翻译提前终止或影响mRNA 的表达。按照ACMG变异分类标准,可归类为致病性变异(Pathogentic)。与患儿临床症状相关的其他基因未发现病理性变异。
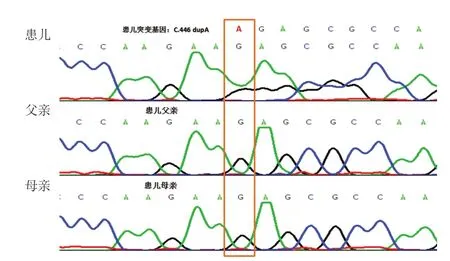
图2 患儿及其父母全外显子基因测序图
根据患儿临床资料及基因分析结果,判定为HIST1H1E基因c.446dupA变异为患儿的致病变异,确诊为RMNS。目前患儿行康复训练,经随访1年,运动能力较前有所好转,语言、智力发育未见明显改善。针对患儿隐睾,已行双侧高位隐睾下降固定术,术后恢复良好。
2 讨论
以Rahman综合征、HIST1H1E基因、组蛋白H1、Rahman syndrome、Histone H1为检索词,分别检索建库至2019年12月中国知网数据库、万方数据库、维普期刊服务平台及PubMed的相关文献。最终检索到符合条件的中文文献,国内尚无RMNS相关报道;相关英文文献4篇[1-4],包含31例RMNS患儿,男13例、女18 例。其中C.430 dupG 为目前已知的RMNS 的热点突变,其次为C.441 dupC、C.435 dupC、C.433 dupG、C.436_458del23。本例患儿c.446dupA变异位点尚未见报道。见表1
HIST1H1E基因定位于人第6 号染色体p 22.2,是一个单外显子基因,无内含子,负责编码具有219个氨基酸、相对分子质量为22 000 的H 1 组蛋白家族成员E[4-5]。含有3 个结构域:中间的球状结构域(Globular domain,GD)、N 末端结构域(N-terminal domain,NTD)和C 末端结构域(C-terminal domain,CTD)[6]。研究发现,CTD末端的净正电荷为高亲和力结合染色质过程所必需,且富含碱性氨基酸,对DNA凝聚起决定作用;当HIST1H1E基因发生突变时,由于HIST1H1E是单外显子基因,逃避了无义介导的RNA衰变,CTD编码区发生移码和提前终止,形成一个截短蛋白,突变蛋白的长度缩短,使其净电荷较野生型蛋白质(净电荷为44)减少了7~9,在结合带负电荷DNA 时能力下降,同时还会阻碍染色质结合和蛋白质之间的相互作用,从而影响人类表观遗传调控过程[1,7-9]。目前已报道的所有RMNS 相关的突变均位于HIST1H1E基因CTD末端的94 个碱基对的区域中,形成相同的具有38 个氨基酸羧基末端的突变蛋白,这是移码突变时阅读框发生相同位移的结果[2]。
RMNS以轻度至重度智力障碍,发育落后和不同程度的躯体过度生长为特征,患儿有相似的面部表现:面部丰满、发际线高、双颞变窄、眦距增宽、眼睑裂狭窄等。本例患儿临床表现与已报道患儿相似。另外,已报道的其他31 例患儿中有19 例在新生儿期有低钾血症;16 例合并行为问题,包括焦虑症、注意力缺陷、多动障碍、自闭症谱系/特征及攻击行为等;15例患儿完善了头颅磁共振成像(MRI),13 例核磁结果异常,以胼胝体异常最常见;13例患儿合并牙齿异常,包括龋齿、牙釉质损伤、牙齿碎裂、牙列不齐等;13例患者有心脏异常,包括房间隔缺损9例,室间隔缺损3例,卵圆孔未闭1例,动脉导管未闭1例;12例骨骼异常:后凸畸形/脊柱侧弯4 例,足弓畸形3 例,下肢不对称2 例,颅骨融合症1 例,远端近臂畸形1 例,多处骨折1例,脚趾重叠1例;13例男孩中9例患有隐睾症;5 例存在甲状腺功能减退症;另有7 例患者有皮肤外胚层异常,包括皮肤痣、头发生长缓慢、斑秃、缺乏体毛和薄指甲等[1-3]。
RMNS 患儿存在不同程度的过度生长[2];且RMNS患儿的身高百分位数有随着年龄的增长而逐渐降低的趋势[3]。表观遗传调控过程破坏是导致过度生长和智力障碍(overgrowth with intellectual disability,OGID)的重要机制。目前发现的可引起OGID的表观遗传调控基因包括NSD1、EZH2、DNMT3A、CDH8、HIST1H1E和EED等。在对710例OGID患儿的研究中发现,致病基因阳性检出率为21.3%,其中NSD 1基因的阳性检出率为17%,HIST1H1E基因的阳性检出率为0.7%[1]。因RMNS 的患儿以智力障碍和不同程度的躯体过度生长为特征,而NSD 1基因突变所致的Sotos 综合征也可表现为躯体过度增长,及智力低下、语言发育落后等中枢神经系统表现,且临床上较RMNS更为常见。本例患儿也曾考虑为Sotos综合征,但本例患儿的外显子分析没有发现其他任何会导致过度生长的变异。同时,RMNS 还应与另一种过度生长综合征Weaver 综合征鉴别。Weaver 综合征也有躯体过度增长、骨龄提前、隐睾、智力障碍等临床表现,全外显子组基因测序有助鉴别诊断。

表1 RMNS患者HIST1H1E基因突变的位点及例数汇总
本例患儿HIST1H1E基因c.446dupA为新发现的变异,编码的氨基酸由丝氨酸变成谷氨酸,经Alamut软件预测可以使氨基酸翻译提前终止或影响mRNA的表达。本例患儿语言运动发育均较同龄儿落后,且有躯体过度生长,特殊面容;血氨基酸及肉碱谱检验异常;骨龄提前,隐睾,智力落后。本例患儿初次就诊时2岁7月龄,身高98 cm(+0.95 SD),目前4岁3月龄,身高107 cm(+0.28 SD)。同时患儿有龋齿。目前暂未发现患儿有明显的眼、甲状腺异常,需定期随访。
综上所述,RMNS 发病率低,且临床症状不典型。对于有轻度至重度智力障碍,发育落后和不同程度过度生长的患儿,必要时需考虑RMNS可能。本例RMNS患儿HIST1H1E基因存在新发现的c.446dupA变异,为人类基因突变数据库提供了新的数据,有助于丰富基因型-表型数据库,扩大表观遗传相关基因在过度生长及智力障碍病因学中的范围。
