含苯并噻唑、喹啉单元荧光材料的合成、结构与光物理性能
2020-06-04陈连清王川川吴忠达
陈连清, 王川川,吴忠达
(中南民族大学 化学与材料科学学院,武汉 430074)
有机发光材料在能源领域具有非常广阔的应用前景,具有发光性能的材料中,一般含有氮杂环如苯并噻唑、喹啉、噁二唑、吡啶、喹喔啉等[1],或含拉电子基团如氟、腈基等.其中苯并噻唑及其衍生物和含喹啉单元化合物最具有代表性.苯并噻唑类化合物具有很强的光学特性[2, 3],比无机发光材料具有发光效率高,颜色选择范围宽等特点,在溶液中的激发光谱和发射光谱存在着很好的对应性,荧光量子产率较高,故被广泛应用于光致发光领域.此前胡景润等[4]将苯并噻唑母体与吡唑琳环相结合进行光物理性能研究,邹迎晖等[5]合成苯并噻唑类化合物的操作步骤较为繁琐,MORA A K[6]等探究溶剂氢键和极性对苯并噻唑在淀粉样原纤维传感器光物理性能方面的影响,然而对苯并噻唑类化合物及其取代基团的发光结构进行修饰调控的研究较少.为此本文尝试采用Jacobson 环化[7]和缩合2-氨基硫酚及苯甲醛衍生物,合成甲氧基取代的2-芳基苯并噻唑类化合物.通过改变所合成的2-芳基苯并噻唑苯环上甲氧基的取代位置和数目,对其发光性能进行改性.利用它们在有机溶剂中的光谱性质来研究结构与发光性能的关系,为探索新型有机发光材料及设计具有特定光电转换功能材料的分子提供依据.
含喹啉单元类化合物,具有良好的成膜性、较高的载流子迁移率及较好的热稳定性[8].其发光材料具有荧光效率高、发光色度纯、载流子传输性好、熔点高、热稳定性较好等优点.侯可宁等[9]将含喹啉单元的双Schiff碱化合物与取代芳胺进行缩合,并与过渡金属的氯化物、硝酸盐、硫酸盐进行配位,制备了一系列发光材料.ZHANG G等[10]以喹啉-8-乙酸乙酯酸和二羧酸为配体与过渡金属(Mn,Zn,Cd或Pb)进行水热配位,得到一系列有机框架化合物.PROSTOTA Y等[11]将吡咯和喹啉进行新型半方酸研究,温宇旸等[12]将吡咯并喹啉酮衍生物进行多组分合成,得到一系列很有趣的发光材料.
然而对含喹啉单元化合物结构的共轭程度进行调控和通过诱导效应改变取代基电子云密度来进行发光改性的研究较少.对此本文尝试通过Friedländer缩合法[13]引入含喹啉单元,并对喹啉衍生物的取代苯环进行调控.如改变其共轭体系大小、引入推拉电子基团等,来合成具有不同光物理性能的喹啉单元化合物,期望喹啉基团的引入能够提高荧光材料的电子传输能力,有利于其捕获电子,使电荷注入平衡,最终达到提高光物理性能的目的.
本文使用Jacobson 环化和缩合2-氨基硫酚及苯甲醛衍生物来合成具有不同发光性质的苯并噻唑类化合物,发现改变其取代基位置和数目对苯并噻唑结构进行修饰调控时,其发光性能够呈现规律性变化.利用Friedländer缩合法,合成具有不同电子诱导效应和大小不同共轭结构的含喹啉单元化合物,探究其发光性质是否发生变化及有何种规律性,测试了这两类目标化合物的紫外-可见、荧光光谱,证明这些化合物具有较好的共轭结构与发光效率,是良好的发光材料.
1 实验部分
1.1 材料和仪器
2-氨基硫酚、甲氧基取代的苯甲醛、对甲基苯磺酸(PTSA)、氯仿、氨气、乙醚、Na2SO4、乙醇、苯乙酮、无水甲苯(氩气保护下经钠钾合金回流后蒸馏纯化);四氢呋喃(氩气保护下经钠钾合金回流后蒸馏纯化)、二氯甲烷(经氢化钙回流干燥后蒸馏纯化);其他药品和试剂均为市售化学纯或分析纯,使用前未经进一步纯化.涉及配合物的反应均在氩气氛围里完成的,进行柱层析的固定相采用200~300目硅胶.
数字显示显微熔点测定仪(X-4型,北京泰克仪器),温度计未校正;核磁共振谱仪(Bruker AVANCE III 400 M型,德国,氘代试剂为CDCl3);傅立叶红外光谱仪(NEXUS-470智能型,美国,KBr压片);元素分析仪(Vario-EL III CHNS型,美国);质谱仪(ZAB 3F-HF型,英国,快原子轰击);紫外-可见光谱分光光度计(Perkin-Elmer Lambda-Bio35,日本津岛);荧光光谱仪(PE LS-55型,美国);单晶衍射仪(Bruker SMART APEXII,德国).
1.2 2-芳基苯并噻唑类化合物的合成
中间产物:对甲氧基苯甲醛(a)、2,4-二甲氧基苯甲醛(b)、2,4,5-三甲氧基苯甲醛(c)参考文献[14-16]进行合成.
利用Jacobson 环化和缩合2-氨基硫酚及苯甲醛衍生物合成目标化合物步骤如下:称取2-氨基硫酚(10 mmol)及甲氧基取代的苯甲醛(10 mmol)及PTSA(1.0 mmol)在氯仿(20 mL)里,通氨气的条件下70 ℃回流24 h.冷却后,混合物用水及乙醚提取,再用Na2SO4干燥.旋蒸移除溶剂后,粗产物用乙醇重结晶得到纯品.合成路线如图1.

图1 产物T1~T5的合成路线
产物T1~T5相关物性常数及表征数据如下:
2-(2-甲氧苯基)-苯并噻唑(T1):浅绿色晶体,产率85%,m.p.118~119 ℃.1H NMR:8.52(d,J= 8.0 Hz, 1H),8.08(d,J= 8.0 Hz, 1H),7.91(d,J= 7.6 Hz, 1H),7.46(m, 2H),7.36(t,J= 7.6 Hz, 1H),7.10(m, 2H),4.06(s, 3H),Anal.calcd for C14H11ONS: C 69.68, H 4.59, N 5.90; found C 69.72, H 4.63, N 6.01.MS(FAB):m/e,241(M+).
2-(4-甲氧苯基)-苯并噻唑(T2):白色晶体,产率86%,m.p.122~123 ℃.1H NMR:8.03(d,J= 7.2 Hz, 3H),7.86(d,J= 8.0 Hz, 1H),7.46(t,J= 7.6 Hz, 1H),7.34(t,J= 7.6 Hz, 1H),7.00(d,J= 8.0 Hz, 2H),3.88(s, 3H).Anal.calcd for C14H11ONS:C 69.68, H 4.59, N 5.80; found C 69.68, H 4.59, N 5.80.MS(FAB):m/e,241(M+).
2-(3-甲氧苯基)-苯并噻唑(T3):白色晶体,产率82%,m.p.120~121 ℃.1H NMR:8.06(d,J= 7.6 Hz, 1H),7.90(d,J= 7.6 Hz, 1H),7.65(m, 2H),7.49(t,J= 7.2 Hz, 1H),7.39(t,J= 7.2 Hz, 2H),7.04(d,J= 7.6 Hz, 1H),3.92(s, 3H).Anal.calcd for C14H11ONS: C 69.68, H 4.59, N 5.80; found C 69.57, H 4.56, N 5.76.MS(FAB):m/e,241(M+).
2-(2,4-二甲氧苯基)-苯并噻唑(T4):黄绿色晶体,产率77%,m.p.139~140 ℃.1H NMR:8.45(d,J= 8.0 Hz, 1H),8.02(d,J= 8.4 Hz, 1H),7.87(d,J= 8.0 Hz, 1H),7.45(t,J= 7.2 Hz, 1H),7.32(t,J= 7.2 Hz, 1H),6.65(d,J= 8.8 Hz, 1H),6.56(s, 1H),4.01(s, 3H),3.86(s, 3H).Anal.calcd for C15H13O2NS:C 66.40, H 4.83, N 5.16; found C 66.32, H 4.76, N 5.02.MS(FAB):m/e,271(M+).
2-(2,4,5-三甲氧基苯基)-苯并噻唑(T5):绿色晶体,产率73%,m.p.198~199 ℃.1H NMR:8.05(t,J= 7.2 Hz,2H),7.88(d,J= 7.6 Hz, 1H),7.45(t,J= 7.6 Hz, 1H),7.33(t,J= 7.2 Hz, 1H),6.61(s, 1H),4.04(s, 3H),4.01(s, 3H),3.96(s, 3H).Anal.calcd for C16H15O3NS:C 63.77, H 5.01, N 4.65; found C 63.66, H 4.96, N 4.62.MS(FAB):m/e,301(M+).
1.3 6-溴-2-芳基-4-苯基-喹啉化合物及衍生物的合成:
中间产物d~h的合成步骤如下:
对氟苯乙酮(d):准确称取对氟苯甲酸(0.57 g, 4.0 mmol)于Schlenk管中,加入26 mL 无水无氧处理的THF,控制温度为-78 ℃,逐滴加入1.6 mol/L的甲基锂乙醚溶液(8.0 mmol, 5.0 mL),缓慢升温至室温,加入1.01 g氯化铵的饱和溶液,乙醚萃取有机相,无水硫酸钠干燥.用展开剂[V(石油醚)∶V(氯仿)=2∶1]柱层析纯化,得目标产物.产品对氟苯乙酮为无色油状液体,称重0.53 g,产率95.3%.
对甲氧基苯乙酮(e):准确称取对甲氧基苯甲酸(0.57 g, 4.0 mmol)于Schlenk管中,加入26 mL无水无氧处理的THF,控制温度为-78 ℃,逐滴加入1.6 mol/L的甲基锂乙醚溶液(8.0 mmol, 5.0 mL),缓慢升温至室温,加入1.01 g氯化铵的饱和溶液,乙醚萃取有机相,无水硫酸钠干燥.用柱层析纯化(展开剂同上),得无色油状液体0.45 g,产率84.5%.
萘乙酮(f):在50 mL三口烧瓶中将无水三氯化铝(1.61 g, 12.0 mmol)加入10 mL 1,2-二氯乙烷中,冰浴下逐滴加入乙酰氯(0.62 mL, 10.5 mmol).再将萘(1.28 g, 10.0 mmol)溶于4.0 mL 1,2-二氯乙烷中,逐滴加入上述溶液,搅拌1 h,放置过夜,将混合物倒入加了少量浓盐酸的50 mL碎冰中,静置,用二氯乙烷萃取,干燥后用柱层析纯化,得无色油状液体0.87 g,产率51.1%.
5-溴-2-氨基-苯甲酮(g):在50 mL烧瓶中,加入5-溴-3-苯基-2,1-苯并异噁唑(0.73 g, 2.7 mmol)溶解于7.0 mL HOAc中,再加入铁粉(1.29 g, 21.6 mmol)和2.5 mL 水,回流2.5 h,冷却过滤,将滤液倒入20 mL水中,用乙醚萃取,稀NaHCO3溶液洗涤,干燥,旋干,粗产品用20 mL甲醇重结晶,得到5-溴-2-氨基-苯甲酮:0.67 g,产率89%,m.p.109~112 ℃(文献值:110~111 ℃).
5-溴-3-苯基-2, 1-苯并异噁唑(h):在50 mL烧瓶中,加入氢氧化钾(4.80 g, 227 mmol)和30 mL甲醇,冰浴下再加入苯乙腈(1.67 mL, 12.6 mmol).将对溴硝基苯(2.54 g, 12.6 mmol)溶于10 mL THF和20 mL甲醇的混合溶液中,逐渐加入上述溶液中,溶液变为深紫色.冰浴下搅拌4.5 h,将混合物倒入100 mL水中,水解过夜.有大量黄色针状晶体生成,过滤,水洗至中性,甲醇重结晶.5-溴-3-苯-2,1-苯并异噁唑:2.34 g,产率80%,m.p.115~118 ℃(文献值:116~117 ℃).
通过Friedländer缩合法合成目标化合物步骤如下:
称取10 mmol 5-溴-2-氨基-苯甲酮和10 mmol的酮衍生物,依次加入50 mL圆底烧瓶中,再加入10 mL 醋酸作为溶剂,加入0.1 mL浓硫酸作为催化剂,氩气保护下,搅拌加热到120 ℃,反应过夜.待反应结束后,将反应液倒入浓氨水溶液里,不停搅拌,溶液中出现大量固体,过滤后,用水冲洗数次,粗产品减压干燥后,采用硅胶柱层析分离,旋蒸干燥得到纯品.合成路线如图2.
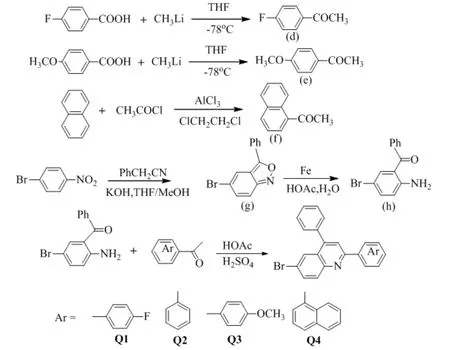
图2 产物Q1~Q4的合成路线
产物Q1~Q4相关物性常数及表征数据:
6-溴-2-(4-氟苯基)-4-苯基-喹啉(Q1):白色固体,产率64%,m.p.214~215 ℃.1H NMR : 7.22(t,J= 8.4 Hz, 2H), 7.48~7.56(m, 6H), 7.76(t,J= 8.8 Hz, 1H), 7.79(s, 1H),7.91(d,J= 8.4 Hz, 1H), 8.20~8.22(m, 3H).Anal.calcd for C21H13BrFN: C 66.68, H 3.46, N 3.70; found C 66.75, H 3.53, N 3.76.MS(FAB):m/e,377(M+).
6-溴-2, 4-二苯基-喹啉(Q2):白色固体,产率67%,m.p.223~224 ℃.1H NMR : 8.18(d,J= 6.4 Hz, 2H), 8.10(d,J= 9.2 Hz, 1H), 8.03(d,J= 6.8 Hz, 1H), 7.78~7.83(m, 2H), 7.47~7.51(m, 8H).Anal.calcd for C21H14BrN: C 70.01, H 3.92, N 3.89; found C 70.12, H 3.86, N 3.94.MS(FAB):m/e,359(M+).
6-溴-2-(4-甲氧基)-4-苯基-喹啉(Q3):淡黄色固体,产率70%,m.p.229~230 ℃.1H NMR : 3.90(s, 3H), 7.06(d,J= 8.4 Hz, 2H), 7.46(td,J= 7.6, 1.2 Hz,1H), 7.53~7.58(m, 5H), 7.72(td,J= 7.6, 1.2 Hz, 1H), 7.79(s,1H), 7.89(d,J=8.4 Hz, 1H), 8.19(d,J= 8.8 Hz, 2H), 8.24(d,J= 8.4 Hz, 1H).Anal.calcd for C22H16BrNO: C 67.71, H 4.13, N 3.59; found C 67.79, H 4.24, N 3.52.MS(FAB):m/e,389(M+).
6-溴-2-萘基-4-苯基-喹啉(Q4):黄色固体,产率61%,m.p.246~247 ℃.1H NMR : 8.20(d,J= 7.6 Hz, 1H), 8.17(s, 1H), 8.11(d,J= 8.8 Hz, 1H), 8.04(d,J=2.4 Hz, 2H), 7.84(s ,1H), 7.81(d,J= 2.4 Hz, 1H), 7.58~7.48(m , 9H).Anal.calcd for C25H16BrN: C 73.18, H 3.93, N 3.41; found C 73.26, H 3.87, N 3.49.MS(FAB):m/e,409(M+).
2 结果与讨论
2.1 合成路线的选择
通过Jacobson 环化和缩合2-氨基硫酚及苯甲醛衍生物合成5种甲氧基取代的2-芳基苯并噻唑化合物.实验以PTSA为催化剂,氯仿为溶剂,取得了理想效果.当2-氨基硫酚和甲氧基取代的苯甲醛摩尔比为1∶1时,反应较为彻底,通氨气回流24 h.冷却后,混合物用水及乙醚萃取,再用Na2SO4干燥,移除溶剂后,用乙醇重结晶即可得到纯品,避免了苯并噻唑的共轭体系与硅胶吸附而难以分离.2-氨基硫酚与甲氧基取代的苯甲醛发生缩合反应,产物产率与苯环上甲氧基的取代数目有关,目标产物T1~T3产率较高,而当甲氧基数目增多时,由于空间位阻效应,T4、T5产率略微下降.5种产物中,因甲氧基取代位置和数目不同,化合物紫外吸收波长逐渐红移,化合物T5呈绿色晶体.
通过Friedländer缩合法对喹啉苯环进行调控合成4种含喹啉单元的6-溴-2-芳基-4-苯基-喹啉衍生物.实验以浓硫酸为催化剂,醋酸为溶剂,取得了理想效果.当5-溴-2-氨基-苯甲酮和酮的衍生物摩尔比为1∶1时反应较为彻底,氩气保护下,搅拌加热到120 ℃.反应过夜,将反应液在浓氨水溶液中不停搅拌,出现大量固体,过滤后,用水冲洗,粗产品减压干燥,硅胶柱层析分离,旋蒸干燥得到纯品.Friedländer法为缩合反应,产物产率取决于诱导效应对苯环上电子云密度的影响,随着苯基引入强推电子基团(—OCH3)时,产率略有增高,Q3产率较高;当合成Q4,取代基换成共轭程度和位阻更大的萘环时,产率出现下降.4种产物中,紫外吸收随着推电子的诱导效应增强和共轭体系增大而逐渐红移.9种化合物的物性常数见表 1.

表1 9种化合物的物性常数
注:a为理论值,b为实测值
2.2 产物的结构表征
表2为9种化合物的核磁氢谱和质谱归属结果.由核磁氢谱归属数据可见:芳环或萘环上δH约为6~8;δH在3.96~4.06时,可归属为—OCH3的H的特征化学位移.表中质谱数据显示了各化合物的分子离子峰.

表2 9种化合物的核磁氢谱和质谱数据
由9种化合物各自的质谱、核磁氢谱和元素分析的数据,证实其与所设计的目标化产物的结构相符.
2.3 产物的光物理性能
2.3.1 紫外-可见光谱
在室温下,5种苯并噻唑类化合物在CH2Cl2溶液中的紫外-可见吸收光谱见图3.由图3可知:5种化合物具有广泛吸收带的区域为293~345 nm, 归属为苯环和噻唑环的共轭吸收峰,摩尔吸光系数约10000 mol-1cm-1.其具有明显的精细结构和较大的共轭体系,主要归结于化合物共轭体系的π-π*跃迁[17].化合物T1的紫外吸收峰为293 nm,当改变甲氧基的取代位置从3位到2位到4位,即化合物从T1(间位取代)到T2(邻位取代)到T3(对位取代)时, 吸收波长明显增加,红移值分别为15 nm和27 nm;当增加取代甲氧基数目从1个到3个,即从化合物T1到T4到T5时,吸收波长依次红移.即在相同条件下,甲氧基在苯环上从间位取代、邻位取代到对位取代,紫外吸收波长依次变长,苯环上甲氧基的数目越多时,相应化合物的紫外吸收波长越长.

图3 苯并噻唑类化合物T1~T5的紫外-可见吸收光谱图
4种喹啉类化合物Q1~Q4的紫外-可见吸收光谱见图4.由图4可见:化合物Q2的紫外吸收峰为304 nm.化合物Q4的吸收峰值比化合物Q2峰值红移了40 nm;化合物Q1的吸收峰值比Q2的吸收峰值蓝移了20 nm,而化合物Q3的吸收峰值比Q2的吸收峰值红移了23 nm.由紫外-可见吸收光谱数据可见:化合物在光照条件下获得能量受到激发,在跃迁到激发态的过程中化合物Q4比化合物Q2吸收波长长,即相同条件下,共轭体系越大的喹啉衍生物吸收波长越长.化合物Q3比化合物Q2吸收波长长,即在相同条件下,含有推电子基的喹啉衍生物吸收峰发生红移. 而化合物Q1比化合物Q2吸收峰蓝移,原因在于含有拉电子基的喹啉衍生物吸收波长要短.即喹啉取代基单元部分引入推电子基团比拉电子基团紫外吸收波长变长,取代单元的共轭体系越大,吸收波长越长.

图4 喹啉类化合物Q1~Q4的紫外-可见吸收光谱图
2.3.2 荧光图谱
由图5可见:化合物T1的荧光发射峰在354 nm.依照顺序3位(间位取代)、2位(邻位取代)、4位(对位取代)改变甲氧基的位置时,观察到荧光发射波长依次红移.当甲氧基数目从1个增加到3个时,即从化合物T1到T4到T5时,荧光发射波长也逐渐变长.在相同条件下,当甲氧基从苯环的间位取代、邻位取代到对位取代,荧光发射波长依次增长,苯环上甲氧基的数目越多时,相应化合物的荧光发射波长越长.
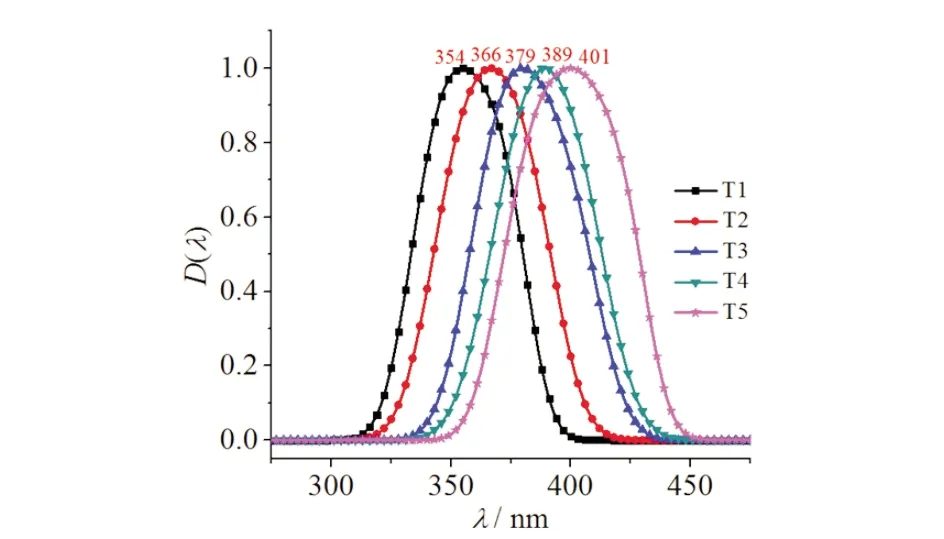
图5 苯并噻唑类化合物T1~T5的荧光光谱图
4种喹啉化合物Q1~Q4的荧光光谱见图6.化合物Q2的荧光发射峰为356 nm.化合物Q4的发射波长比化合物Q2的发射峰值红移了33 nm.化合物Q1的发射波长比Q2的发射峰值蓝移了12 nm,而化合物Q3的发射波长比Q2的发射峰值红移了22 nm.从荧光光谱数据可以看出,在化合物在从激发态回到基态的过程中,化合物Q4比化合物Q2发射波长长,即相同条件下,化合物的共轭体系越大,其发射波长越长,而含有推电子基的喹啉衍生物发射波长变长. 化合物Q1比化合物Q2发射波长要短,即在相同条件下,含有拉电子基的喹啉衍生物荧光发射波长变短.即喹啉取代基单元部分引入推电子基团比拉电子基团使荧光发射波长变长,取代单元的共轭体系越大,发射波长越长
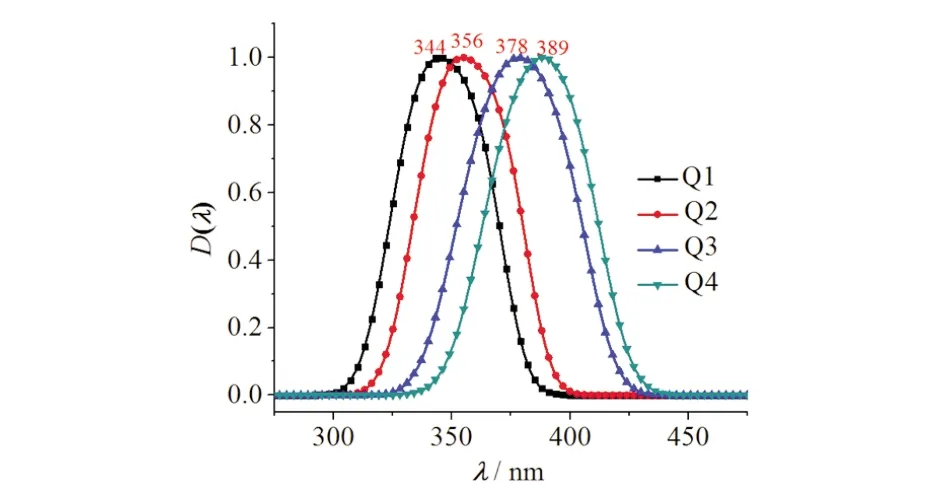
图6 喹啉类化合物Q1~Q4的荧光光谱图
9种化合物的紫外-可见吸收和荧光发射数据总结如表3所示.数据表明所有化合物结构进行修饰后,其光物理性能可变可控,改变化合物T1到T5上甲氧基的取代位置和个数,其紫外可见吸收和荧光发射波长都发生规律性改变.甲氧基在苯环上的取代位置从间位到邻位到对位,紫外可见吸收波长和荧光发射波长依次红移,取代的甲氧基的数目越多,波长红移越多.而取代基的种类不同、诱导效应不同、化合物的共轭程度大小不同也会影响其光物理性能.改变化合物Q1到Q4的取代基种类和共轭程度,其紫外可见吸收和荧光发射波长也发生了规律性改变.不同取代基团推拉电子的诱导效应不同,从具有拉电子效应的对氟苯基到苯基,再到具有推电子效应的对甲氧基苯基,苯环上电子云密度逐渐增大,相应化合物的紫外可见吸收和荧光发射波长逐渐红移.当换成具有更大共轭效应的萘环进行取代时,波长进一步红移,即共轭体系越大的喹啉衍生物,发射波长越长.因为含推电子基团的荧光体激发态常由环外甲氧基上的电子激发转移到环上而产生,其电子云几乎与芳环上的轨道平行,实际上共享了共轭电子结构,同时扩大了其共轭双键体系,甲氧基数目更多,影响效果更大,所以其吸收波长与发射波长均比未被取代化合物的波长长,其荧光效率增加.而含F苯基从反面证明了拉电子基团所引起的n-π 跃迁是禁阻跃迁,结果是S1-T1系间窜越过程被加强,导致了荧光减弱的现象.喹啉类化合物中萘基的数据表明:不仅取代基电子效应对光物理性能有影响,直接换成更大共轭体系,共轭程度更大,紫外可见吸收和荧光发射波长进一步红移.共轭体系具有大的π键结构,共轭体系越大,离域电子越容易被激发,相应地紫外吸收和荧光较易产生.荧光量子产率数据表明:该两类化合物具有较好的荧光量子产率,是较好的发光材料.
表3 9种化合物的紫外-可见吸收和荧光发射数据
注:a在298.3 K的二氯甲烷溶液中;b在5.0%重量比的PMMA薄膜中;c相对于硫酸奎宁(在0.5 mol/L H2SO4中10-5mol/L,Φf= 0.546,作为标准),在CH2Cl2溶液中测量荧光的量子产率
2.4 晶体结构
化合物2-(2,4,5-三甲氧基苯基)-苯并噻唑(T5)的分子结构图和和堆积结构图分别见图7和图8.T5的晶胞参数和部分键长、键角数据分别见表4和表5.在晶体结构中,五元噻唑环几乎与其稠合苯环共平面,并且两个环平面之间的二面角仅为0.3(1)o.由于空间位阻,苯并噻唑环略微偏离C13—C8苯环的平面[18].它们之间的二面角是4.5(2)o.由于来自相邻苯并噻唑部分的空间干扰,2-甲氧基被扭曲出C13—C8苯平面.C10—C9—O1—C16扭转角为4.8(3)°,其他两个甲氧基几乎位于C8—C13苯平面上.C7—N1[1.301(2)Å]和C1—N1[1.387(2)Å]的键距平均趋势,这是π电子离域的结果.类似地,C6—S1[1.7292(19)Å]和C7—S1[1.7588(17)Å]的键长具有小的差异.
在晶体堆积中,两个相邻分子之间存在苯环的有效重叠.最短的间隔距离约为3.502 Å,足够短以表明π-π堆叠相互作用.C15...H与相邻分子的苯环之间也存在相互作用,最短距离为3.494 Å.分子间相互作用对其光致发光性质具有直接影响,这与其共轭结构的影响相一致.
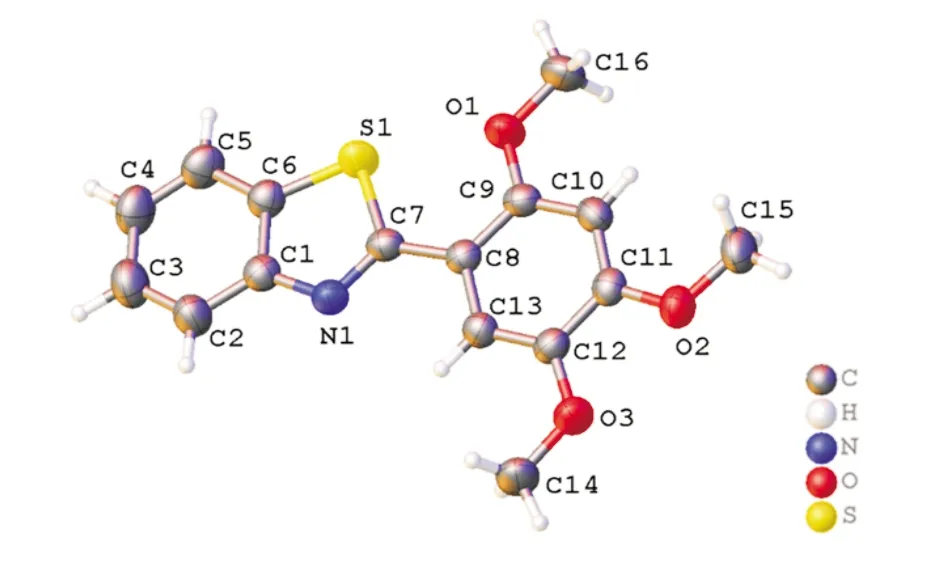
图7 2-(2,4,5-三甲氧基苯基)-苯并噻唑(T5)的分子结构图
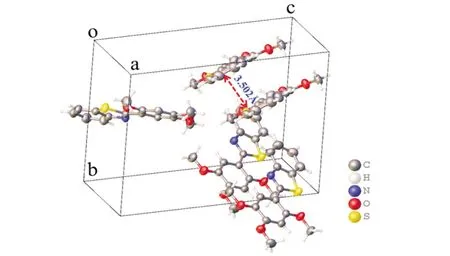
图8 2-(2,4,5-三甲氧基苯基)-苯并噻唑(T5)的堆积结构图
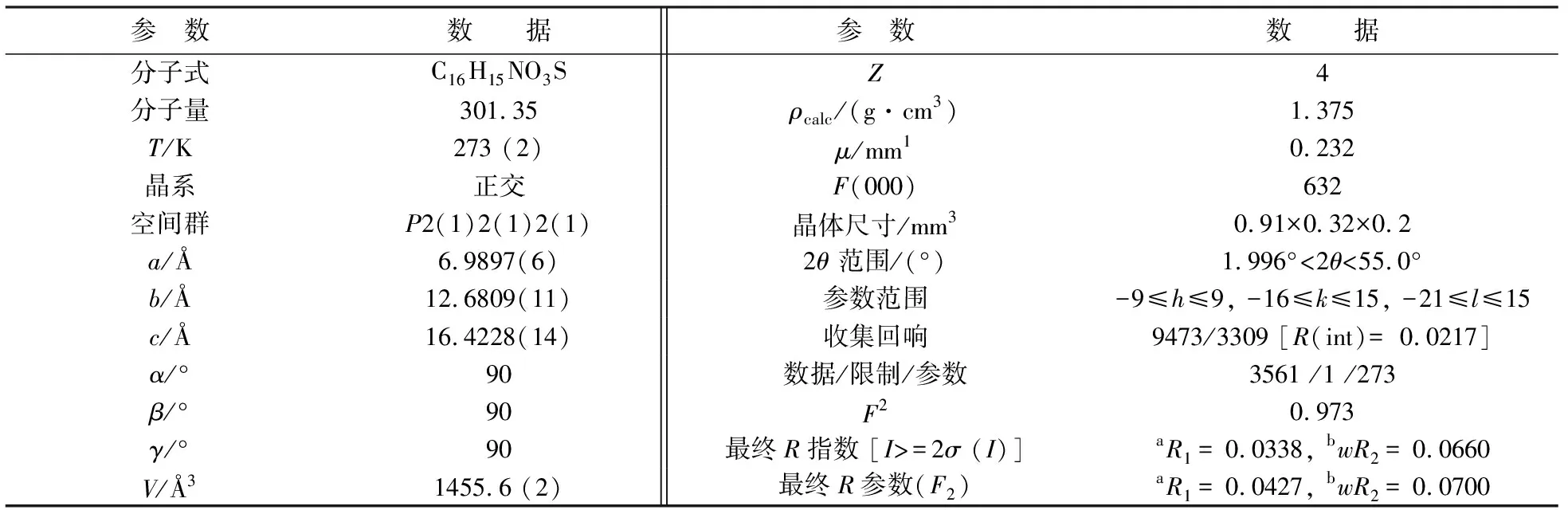
表4 化合物T5的晶体数据和结构细化
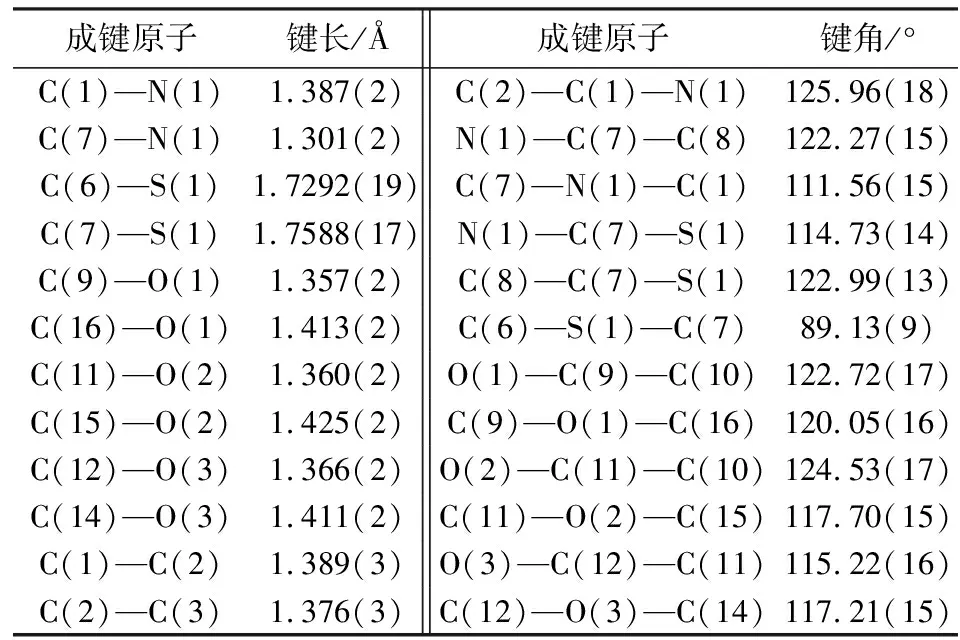
表5 化合物T5选择的部分键长(Å)和键角(°)
3 结语
通过Jacobson 环化和缩合2-氨基硫酚及苯甲醛衍生物合成了5种甲氧基取代的2-芳基苯并噻唑T1~T5;通过Friedländer缩合法对喹啉苯环进行有目的的调控合成了4种含喹啉单元的6-溴-2-芳基-4-苯基-喹啉化合物Q1~Q4.对9种化合物进行了1H NMR、质谱、元素分析等表征,通过单晶衍射确证了2-(2,4,5-三甲氧基-苯基)-苯并噻唑(T5)的晶体结构,测试了这些化合物的紫外-可见、荧光光谱,探讨了其光物理性能.结果表明:化合物特定取代基的位置和个数会影响其光物理性能; 取代基的种类不同、诱导效应不同、化合物的共轭程度大小不同会影响化合物的光物理性能.改变2-芳基苯并噻唑苯环上甲氧基的数目和取代位置,改变喹啉单元的共轭体系大小、引入推拉电子基团,其发光性能具有可变可控的规律性.得到的这两类化合物的结构与设计的目标产物结构相符合,且具有结构精细、共轭效应良好、发光效率高的特点,为设计合成具有优良发光性能的有机发光材料奠定一定基础.
