基于LC-MS/MS 法分析生、炙甘草中水溶性成分
2020-06-02段伟萍李缘嫒郑云枫李存玉陆兔林陈丽红彭国平
段伟萍李缘嫒郑云枫 李存玉 陆兔林陈丽红彭国平
(1.南京中医药大学药学院,江苏南京210023; 2.江苏省中药资源产业化过程协同创新中心,江苏 南京210023; 3.南京中医药大学国家教育部中药炮制规范化及标准化工程研究中心,江苏 南京210023)
甘草为豆科植物甘草Glycyrrhiza uralensisFisch.、胀果甘草Glycyrrhiza inflataBat.或光果甘草Glycyrrhiza glabraL.的干燥根及根茎,是最常用的中药之一[1-2]。历代以来,甘草的临床应用以生、炙甘草为主,其中炙甘草是由生甘草拌入炼蜜后加热炮制而来[3],具有补脾和胃、益气复脉之功效[1]。考虑到中药炮制功效的改变与化学成分变化存在着密切的关系,有研究采用HPLC 指纹图谱技术对甘草蜜炙前后色谱图进行比较,发现生、炙甘草中一些色谱峰面积存在差异[4-5];也有文献对甘草蜜炙前后甘草苷、甘草酸等主要指标性成分进行定量分析研究,结果显示在炮制过程中成分出现了增减变化[6-7]。
本研究在前期研究的基础上[8-10],采用LCMS/MS 定性鉴别结合Marker View 软件分析,系统解析了生、炙甘草水溶性差异成分,进一步以HPLC 梯度波长法检测了炮制前后黄酮及三萜皂苷水溶性成分的含有量变化,以期为炙甘草的炮制及质量评价提供参考。
1 材料
AB SCIEX Triple TOFTM5600 质谱仪,电喷雾离子源(ESI),Analyst TF 1.6 和Peak View 1.2 软件(美国AB 公司);Agilent 1100 高效液相色谱仪(美国Agilent 公司);JCS-1000 电子天平(凯丰集团有限公司);AL204 电子分析天平(万分之一,瑞士梅特勒-托利多仪器有限公司);BT125D 分析天平(十万分之一,德国赛多利斯科学仪器有限公司)。
对照品芹糖甘草苷 (JBZ180106-139,纯度98.1%)、芹糖异甘草苷 (JBZ180205-813,纯度98.5%)、异甘草 苷 (JBZ180314-015,纯 度98.3%) 均购自南京金益柏生物科技有限公司;对照品甘草苷(111610-201607,纯度93.1%)、甘草酸铵(110731-201619,纯度97.0%) 均购自中国食品药品检定研究院;对照品22β-乙酰甘草酸、甘草皂苷G2、乌拉尔皂苷B,均由实验室自制,经HPLC 分析,纯度≥99.0%。乙腈、甲酸均为色谱纯(德国Merck 公司),水为Milli-Q 超纯水,其他试剂均为分析纯。
6 批生甘草饮片购自内蒙古亿利资源集团有限公司甘草分公司(批号20181101~20181106)。经南京中医药大学鉴定教研室严辉副教授鉴定为豆科植物甘草Glycyrrhiza uralensisFisch.的干燥根和根茎。
2 方法与结果
2.1 炙甘草制备 蜜炙甘草,取炼蜜(100 g 生甘草用炼蜜25 g) 加少量水稀释,加入生甘草饮片中拌匀,稍焖,用文火炒至深黄色、透香气、不粘手时取出放凉。
2.2 供试品溶液制备 取生甘草或炙甘草样品粉末(过3 号筛) 约0.5 g,精密称定,加入水25 mL,置100 mL 圆底烧瓶,称定质量,加热回流1 h,放至室温,补足减失质量,滤过,精密移取续滤液5 mL,置10 mL 量瓶中,甲醇定容至刻度,摇匀,离心(8 000 r/min,10 min) 取上清液,即得。
2.3 对照品溶液制备 取8 种对照品适量,精密称定,加50%甲醇溶解,摇匀,制成芹糖甘草苷、甘草苷、芹糖异甘草苷、异甘草苷、22β-乙酰甘草酸、甘草皂苷G2、甘草酸、乌拉尔皂苷B 质量浓度分别272.20、231.71、67.50、86.00、246.68、242.24、715.56、92.00 mg/mL 的混合 对照品溶液。
2.4 色谱条件 Hedera C18色谱柱 (4.6 mm×250 mm,5 μm);流动相乙腈 (A) -0.2% 甲酸(B),梯度洗脱(0~15 min,19%~25% A;15~25 min,25%~30% A;25~41 min,30%~45% A,41~ 60 min,45%~ 70% A);体积流 量1.0 mL/min;柱温25 ℃;0~15 min 在276 nm 下检测芹糖甘草苷和甘草苷,15~25 min 在360 nm下检测芹糖异甘草苷和异甘草苷,25~60 min 在254 nm 下检测22β-乙酰甘草酸、甘草皂苷G2、甘草酸和乌拉尔皂苷B;进样量10 μL。
2.5 质谱条件 电喷雾离子源(ESI);正、负离子检测模式;一级质谱条件为扫描范围m/z50~1 500;毛细管温度550 ℃;离子喷雾空载电压5 500 V/-4 500 V,解簇电压100 V/-100 V,碰撞能量10 V/-10 V,喷雾电压5 500 V,解簇电压100 V;二级质谱条件为扫描范围m/z50~1 500,解簇电压100 V/-100 V,碰撞能量35 V/-35 V,碰撞能量幅度-20 V,雾化器60 psi (1 psi =6.985 kPa),辅助加热器60 psi,气帘气40 psi。
2.6 定性分析
2.6.1 差异性化合物分析 按“2.2” “2.3” 项下方法制备供试品和对照品溶液。采用正、负离子模式分别进行LC-MS/MS 检测,结果显示,在负离子模式条件下生、炙甘草中黄酮、三萜皂苷类成分的质谱响应值较高,见图1。

图1 生、炙甘草水溶性成分LC-MS/MS (-) TIC Fig.1 LC-MS/MS (-) TIC of water-soluble components from raw and honey-roasted licorice
依据LC-MS/MS 所测样本保留时间、各色谱峰离子响应强度和二级质谱信息,在 Marker View1.2.1 软件中设置质谱处理参数为最小保留时间3.0 min,最大保留时间60.0 min,最小质荷比宽度10×10-6,最小保留时间宽度6 scans,噪波阈值100,保留时间容差16 min,质荷比容差1×10-6[11],最大峰数目500,选择色谱峰响应值≥e3,去除同位素峰;进一步将生、炙甘草两组LCMS/MS 总离子流图导入Marker View 软件中,对两组数据进行t检验,获得差异性成分的Log (Fold change) versus p-value 图,见图2,图中每个散点代表一种差异性成分,其位置离X 轴越远,代表该成分在生、炙甘草样本中的差异性越大;以∣Log (Fold change) ∣≥0.10 为界值,筛选出了33种显著的差异性成分,见图1。
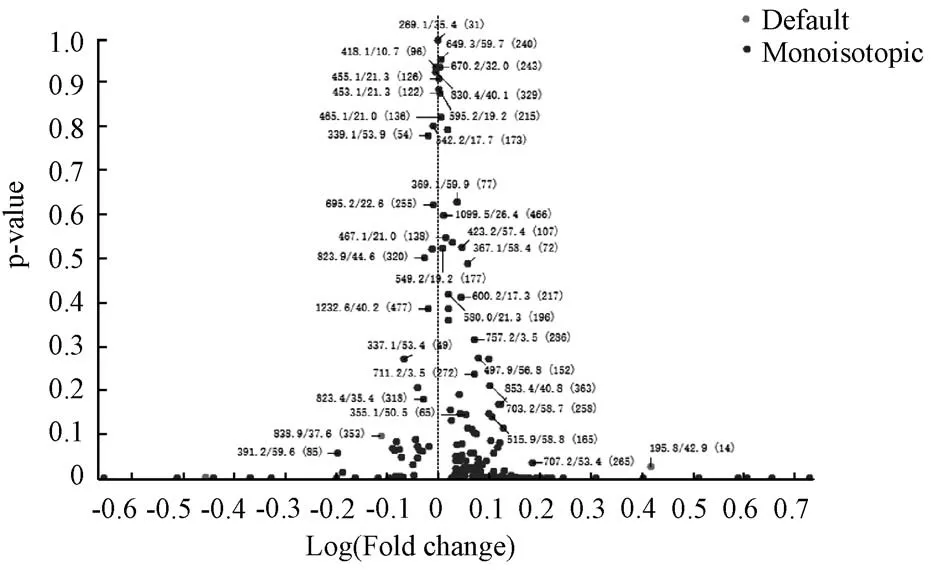
图2 生、炙甘草差异性成分Log (Fold change)versus p-value 图Fig.2 Log (Fold change) versus p-value of different components of raw and honey-roasted licorice
2.6.2 差异性成分定性鉴别 依据相关数据库Chemspider、Scifinder 检索以及甘草化学成分研究报道,建立甘草化学成分数据库,进而对各主要差异性成分的质谱信息进行识别与矫正,推测化合物结构。其中,峰2,4,7,17,19,22 和24 由对照品对照进行了确认,分别为芹糖甘草苷、甘草苷、异甘草苷、22β-乙酰甘草酸、甘草皂苷G2、甘草酸和乌拉尔皂苷B。其他成分依据已建立的目标数据库,通过分子式匹配以及化合物质谱裂解碎片信息进行结构鉴定[12-16]。其中,甘草黄酮类成分在负离子模式下碎片信息丰富,常见特征分子量丢失 为15 Da (CH3)、18 Da (H2O)、32 Da(OCH3)、56 Da ((CH3)2CH =CHCH3)、132 Da(芹糖)、162 Da (葡萄糖) 等;而三萜皂苷成分在负离子模式下碎片信息较少,而在正离子质谱中可见糖取代基分子量丢失132 Da (木糖)、146 Da(鼠李糖)、162 Da (葡萄糖)、176 Da (葡糖糖醛酸),以及苷元上取代基的特征分子量丢失30 Da(C24-OH)、36 Da (2×18,C22-OH)、46 Da (C20-COOH)、60 Da (C22-OAc) 等。
以峰16 为例,在正离子模式下该化合物准分子离子峰为m/z825.425 2 [M+H]+,确定其分子式为C42H64O16,提示该化合物可能为Uralsaponin C;此外,在峰16 的二级质谱中可见2 个连续脱去葡萄糖醛酸基的碎片离子m/z649.393 6 [M+HC6H8O6]+和473.361 7 [M+H-2C6H8O6]+,确定结构中有2 个葡萄糖醛酸取代基,同时从苷元离子连续脱去2 分子H2O 可见碎片离子m/z437.361 7[M+H-2C6H8O6-2H2O]+,表明苷元结构22 位上有羟基取代,以上结构鉴定信息与Uralsaponin C 相一致,最终峰16 鉴定为Uralsaponin C。依据以上差异性成分定性鉴定方法,共推测鉴定了30 种成分,包括12 种黄酮及18 种三萜皂苷,相关质谱信息及化合物结构分别见表1、图3。

表1 甘草蜜炙前后差异性成分定性鉴别Tab.1 Qualitative identification of the differential ingredients between raw and honey-roasted licorice
2.7 含有量变化分析 为了更准确地评价炮制前后主要特征差异性成分的含有量变化,依据LCMS/MS 数据及Marker View 软件分析,选择了4 种黄酮类成分为芹糖甘草苷 (Q-LQ)、甘草苷(LQ)、芹糖异甘草苷(Q-ILQ)、异甘草苷(ILQ)及4 个三萜皂苷类成分为22β-乙酰甘草酸(22β-AGA)、甘草皂苷G2 (LS-G2)、甘草酸 (GA)、乌拉尔皂苷B (US-B) 为指标,采用HPLC 梯度波长进行了含有量测定与对比分析,见图4。

图3 差异性成分结构Fig.3 Chemical structure of differential components
2.7.1 方法学考察
2.7.1.1 精密度试验 按“2.2” 项下方法制备供试品溶液,在“2.4” 项色谱条件下重复进样6次,测得4 种黄酮和4 种三萜皂苷的色谱峰保留时间和峰面积RSD 均小于2.01%,表明仪器精密度良好。
2.7.1.2 稳定性试验 按“2.2” 项下方法制备供试品溶液,在“2.4” 项色谱条件下,分别于0、2、4、6、12、24 h 进样,测得4 种黄酮和4种三萜皂苷的色谱峰保留时间和峰面积RSD 均小于2.44%,表明供试品溶液在24 h 内稳定性良好。
2.7.1.3 重复性试验 精密称定同一批次样品6份,按“2.2” 项下方法制备供试品溶液,在“2.4” 项色谱条件下分别进样,测得4 种黄酮和4种三萜皂苷的色谱峰相对保留时间和相对峰面积的RSD 均小于1.80%,表明该方法重复性良好。
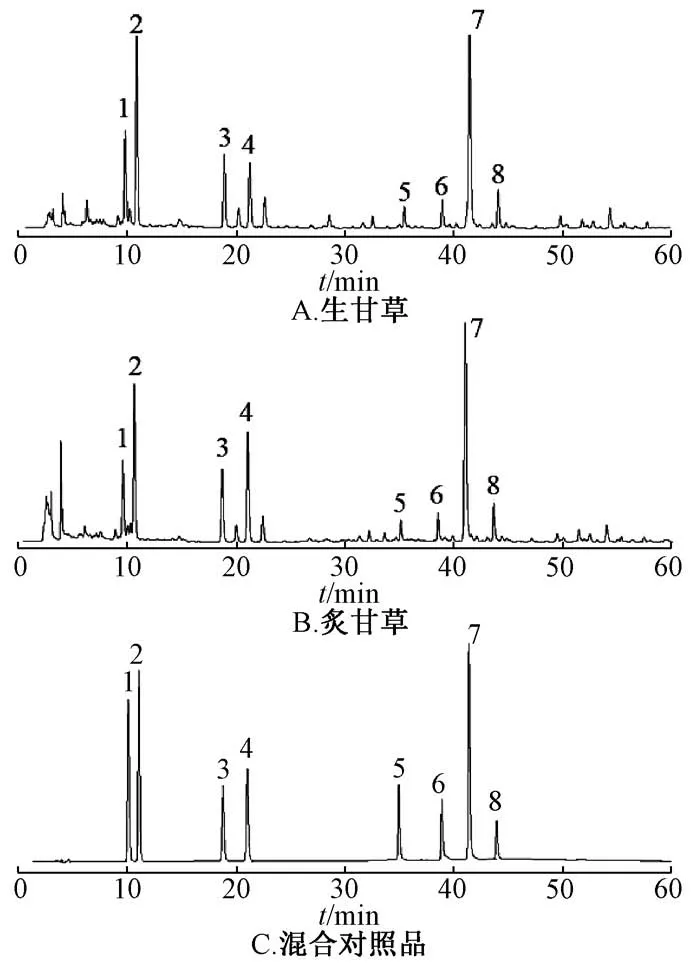
图4 各成分HPLC 色谱图Fig.4 HPLC chromatograms of various constituents
2.7.1.4 线性关系考察 取8 种对照品混合溶液适量,逐级稀释成系列浓度的混合对照品溶液,分别吸取10 μL,在“2.4” 项色谱条件下进样,以峰面积为纵坐标(Y),各对照品质量浓度为横坐标(X),进行回归,结果见表2。表明各成分在各自范围内线性关系良好。

表2 各成分线性关系Tab.2 Linear relationships of various constituents
2.7.2 差异性特征成分定量分析 分别取6 批生甘草及6 批炙甘草,按“2.2” “2.4” 项下方法分别制备供试品溶液并进行测定,以标准曲线计算8种成分的含有量,采用GraphPad Prism 7.0 软件进行单因素方差分析。结果显示,生甘草在蜜炙后芹糖甘草苷、甘草苷、22β-乙酰甘草酸、甘草皂苷G2、甘草酸和乌拉尔皂苷B 成分均下降 (P<0.01);异甘草苷含有量升高(P<0.05),芹糖异甘草苷变化无统计学意义(P>0.05),见图5。

图5 生、炙甘草中差异性特征成分含有量比较Fig.5 Comparison of different characteristic components in raw and honey-roasted licorice
考虑到生甘草炮制过程中加入的炼蜜会对炙甘草中成分的实际含有量产生影响,参考文献[17]以每100 g 炙甘草中含干燥蜂蜜量12 g 计,扣除蜂蜜质量计算炙甘草各成分实际含有量。结果显示,黄酮类成分芹糖异甘草苷和异甘草苷平均含有量分别上升5.11%、25.94%,甘草苷和芹糖甘草苷含有量分别下降21.77%、18.43%,而甘草酸等4 种三萜皂苷成分平均含有量均出现下降,幅度在10.07%~16.69%,结果见表3。
表3 生、炙甘草中各成分含有量变化(,n=6,mg·g-1)Tab.3 Content change of various constituents in raw and honey-roasted licorice (,n=6,mg·g-1)

表3 生、炙甘草中各成分含有量变化(,n=6,mg·g-1)Tab.3 Content change of various constituents in raw and honey-roasted licorice (,n=6,mg·g-1)
3 讨论
传统中医认为,甘草经蜜炙后功效“由清转补”,补脾和胃、益气复脉功效增强,有研究表明,甘草蜜炙后补脾益气药效作用增强,且明显优于生甘草及清炒品[18]。本实验通过开展蜜炙对甘草化学成分的影响研究,发现生、炙甘草水溶性化学成分存在一定的差异,具体表现为,甘草蜜炙后芹糖异甘草苷和异甘草苷含有量上升,芹糖甘草苷和甘草苷含有量下降,基于黄酮成分结构转化规律,应为二氢黄酮成分芹糖甘草苷和甘草苷在炮制加热过程中B 环开环转化为芹糖异甘草苷和异甘草苷所致,有文献表明异甘草苷等甘草黄酮类成分具有抗心律失常[19-21]、保护胃黏膜[22]等作用。炙甘草“益气复脉” 功效,主要表现为增强抗心律失常作用,因此异黄酮类成分的转化及含有量提升,可能与炙甘草益气复脉功效的增强有关。
考虑甘草生、炙品在传统临床使用、配方颗粒和经典名方开发均以水为溶剂进行提取或制备,且生、炙甘草中主要活性成分黄酮和皂苷均具有一定的水溶性,因而本实验选择水回流提取的方式。此外,在含有量测定过程中采用二极管阵列检测器对检测波长进行了考察,结果显示芹糖甘草苷和甘草苷等二氢黄酮类成分在276 nm 波长条件下有较大吸收,芹糖异甘草苷和异甘草苷成分在360 nm 波长条件下吸收较强,而三萜皂苷类成分如甘草酸等在254 nm 波长吸收强度最强,因此本实验选择了梯度波长进行检测。
本实验研究方法能够快速、系统地分析甘草蜜炙前后黄酮及三萜皂苷的成分变化,而差异性组分的解析可为炙甘草炮制及质量标准研究提供一定的依据。但甘草中除了黄酮及三萜皂苷类成分外,还含有多糖及寡糖类组分[23],辅料蜂蜜也含有果糖、葡萄糖等成分[24],在甘草蜜炙过程中糖类组分如何发生变化,尚待后续作进一步探索。
